



Содержание
Перейти к:
https://doi.org/10.20996/1819-6446-2023-03-06
Перейти к:
Пациентка 64 лет с наследственной геморрагической телеангиэктазией (НГТ) госпитализирована в связи декомпенсацией сердечной недостаточности (СН). НГТ диагностирована в 20 лет, рецидивирующие носовые кровотечения – с 52 лет. В 60 лет, после массивного кровотечения, появились симптомы СН. Получала диуретики, препараты железа с эффектом. После объемных кровотечений усугублялась СН. Последнее значимое кровотечение перед настоящей госпитализацией (гемоглобин 67 г/л). При эхокардиографии фракция выброса левого желудочка сохранна, однако присутствовала высокая легочная гипертензия. Данных за тромбоэмболию легочной артерии получено не было, но выявлено интерстициальное поражение легких. В результате пульс-терапии глюкокортикоидами положительной динамики не отмечено, что позволило отвергнуть самостоятельное интерстициальное заболевание легких. В результате лечения СН, а также коррекции анемии состояние пациентки улучшилось. Назначена терапия мацитентаном, от которой пациентка воздержалась. Спустя год больная умерла от острого нарастания легочной гипертензии. По данным литературы, легочная гипертензия при НГТ может оказывать существенное влияние на прогноз и требует своевременной диагностики и лечения. Интерстициальное поражение легких является проявлением основного заболевания и не требует отдельного лечения.
Лутохина Ю.А., Благова О.В., Савина П.О., Заклязьминская Е.В. Болезнь Рандю-Ослера-Вебера с высокой легочной гипертензией и интерстициальным поражением легких. Рациональная Фармакотерапия в Кардиологии. 2023;19(2):179-185. https://doi.org/10.20996/1819-6446-2023-03-06
Lutokhina Yu.A., Blagova O.V., Savina P.O., Zaklyazminskaya E.V. Rendu-Osler-Weber Disease with High Pulmonary Hypertension and Interstitial Lung Disease. Rational Pharmacotherapy in Cardiology. 2023;19(2):179-185. (In Russ.) https://doi.org/10.20996/1819-6446-2023-03-06
Болезнь Рандю-Ослера-Вебера (наследственная геморрагическая телеангиэктазия, НГТ) – это наследственное заболеваниеc аутосомно-доминантным типом наследования, характеризующееся поражением микроциркуляторного русла с развитием телеангиэктазий на коже и слизистых, а также артериовенозных мальформаций в легких, печени и головном мозге [1]. Распространенность этого заболевания составляет порядка 1:6000, в связи с чем НГТ входит в перечень орфанных заболеваний (ORPHA:774) [2][3]. В большинстве случаев (90%) заболевание обусловлено мутациями в генах ENG (эндоглин) или ACVRL1 (костный морфогенетический белок), также описаны мутации в более редких генах SMAD4 (фактор транскрипции Smad4) и GDF2 (костный морфогенетический белок 9 типа) [2]. Мутации в любом из этих генов приводят к нарушению каскада, ответственного за ангиогенез капилляров [4]. Основное проявление заболевания – рецидивирующие носовые кровотечения, приводящие к анемизации, в ряде случаев кровопотерю усугубляют кровотечения из телеангиэктазий, расположенных на слизистой пищевода и желудка [5]. Артериовенозные мальформации в большинстве случаев бессимптомные, однако у ряда пациентов могут приводить к осложнениям, в зависимости от локализации (геморрагический инсульт, образование абсцессов, кровоизлияния) [3].
Помимо классических симптомов заболевания, описаны редкие проявления, такие как легочная гипертензия, которая встречается менее чем у 2% пациентов с НГТ [3]. В данном клиническом случае речь пойдет еще о более редком варианте болезни Рандю-Ослера-Вебера с тяжелой легочной гипертензией и интерстициальным поражением легких.
Информированное согласие. От пациентки и её законного представителя (брата) получено письменное добровольное информированное согласие на публикацию описания клинического случая 19.09.2019 и 02.09.2020 г.
Пациентка Т., 64 лет, поступила в отделение кардиологии Факультетской терапевтической клиники им. В.Н. Виноградова 3.09.2019 г. с жалобами на одышку при небольших нагрузках, отеки нижних конечностей.
На рис. 1 схематически представлен анамнез заболевания пациентки. Известно, что семейный анамнез отягощен по НГТ (у отца и деда пациентки). Болезнь Рандю-Ослера-Вебера дебютировала в 20 лет, когда на коже лица и губ появились телеангиэктазии диаметром до 2-3 мм. Первое клинически значимое носовое кровотечение состоялось в 29 лет, после чего была длительная ремиссия. В течение последних 12 лет носовые кровотечения беспокоят регулярно, в связи с чем страдает железодефицитной анемией и получает внутривенные препараты железа, т.к., со слов пациентки, таблетированные препараты не эффективны. С 43 лет отмечает повышение АД максимально до 190 и 110 мм рт.ст., адаптирована к АД 110- 120/80 мм рт.ст. Получает антигипертензивную терапию, на фоне которой сохраняются эпизоды повышения АД до 150/100 мм рт.ст. При повышении систолического АД >130 мм рт.ст. возникают кровотечения из носа. Ухудшения состояния 2-3 раза в год, когда на фоне дестабилизации АД учащаются носовые кровотечения и усугубляется анемия. С 2016 г., после очередного массивного кровотечения, отметила появление одышки, отеков, асцита. Получала диуретическую терапию, препараты железа на фоне чего самочувствие улучшилось, однако с этого времени после объемных кровотечений усугублялись явления сердечной недостаточности, которые в последующем регрессировали по мере коррекции анемии. Последнее значимое ухудшение состояния, потребовавшее стационарного лечения, в июле-августе 2019 г., когда состоялось 4 профузных носовых кровотечения (08.07, 18.07, 25.07, 02.08), приведшие к значительной кровопотере. На этом фоне гемоглобин составлял 67 г/л. Проводилась инфузионная терапия препаратами железа и гемостатиками, на фоне чего достигнуто повышение уровня гемоглобина до 90 г/л. После выписки сохраняются одышка при минимальных физических нагрузках, а также в покое в положении лежа, отёки голеней и стоп. В этой связи госпитализирована в отделение кардиологии Факультетской терапевтической клиники.
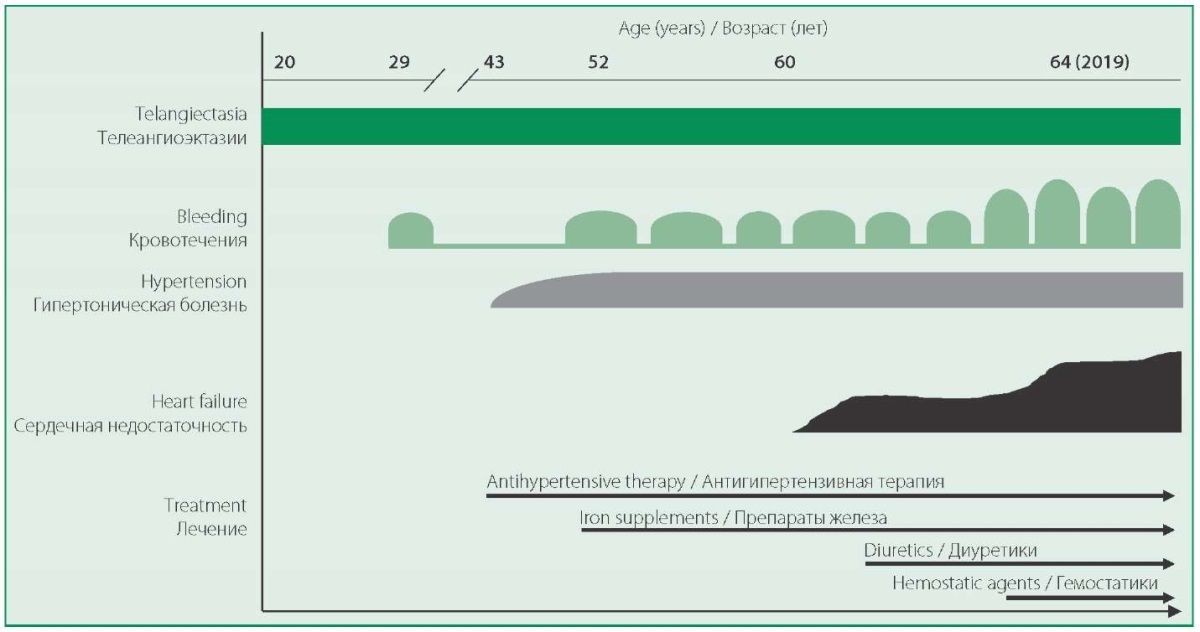
Figure 1. Medical history of patient T.
Рисунок. 1. Анамнез заболевания пациентки Т.
При поступлении состояние пациентки средней степени тяжести. Рост 155 см, вес 78 кг, индекс массы тела 32,47 кг/м2. Температура тела 36,4ºС. Кожные покровы и видимые слизистые бледные, на коже лица, губ, пальцев рук определяются телеангиэктазии диаметром до 2-3 мм. Отеки нижних конечностей до нижней трети бедер. В легких дыхание жесткое, хрипы не выслушиваются, частота дыхательных движений 16/мин, SpO2 87%. Тоны сердца ритмичные, приглушены, частота сердечных сокращений (ЧСС) 68 уд/мин, АД 130 и 80 мм рт.ст. Живот мягкий, безболезненный. Печень на 3,5 см выступает из-под края реберной дуги. Селезенка не увеличена.
В общем анализе крови при поступлении имеются признаки микроцитарной гипохромной анемии легкой степени (гемоглобин 93 г/л, среднее содержание гемоглобина в эритроците 21,7 пг, средняя концентрация гемоглобина в эритроците 285 г/л, средний объем эритроцита 75,9 фл, признаки анизоцитоза, цветовой показатель 0,65, скорость оседания эритроцитов 11 мм/ч). С-реактивный белок 3 мг/л. Биохимический анализ крови, липидный спектр – без значимых отклонений от нормы, за исключением снижения уровня железа до 3,2 мкмоль/л (уровень ферритина не определялся в связи с отсутствием реактивов в лаборатории). Тиреоидные гормоны в пределах референсных значений. Коагулограмма, включая Д-димер, без отклонений от нормы. Общий анализ мочи, анализ кала – без особенностей.
На электрокардиограмме (рис. 2) ритм синусовый, ЧСС 72/мин. Вертикальное положение электрической оси сердца. Интервал PQ 170 мс, QRS 80 мс, QT/QTc 370/405 мс, признаки гипертрофии правого желудочка, ST на изолинии. При суточном мониторировании ЭКГ определялся синусовый ритм, зарегистрировано 115 суправентрикулярных экстрасистол, 427 желудочковых экстрасистол, в т.ч. 1 куплет, 2 пробежки медленной желудочковой тахикардии из 4 QRS с ЧСС до 90 уд/мин.
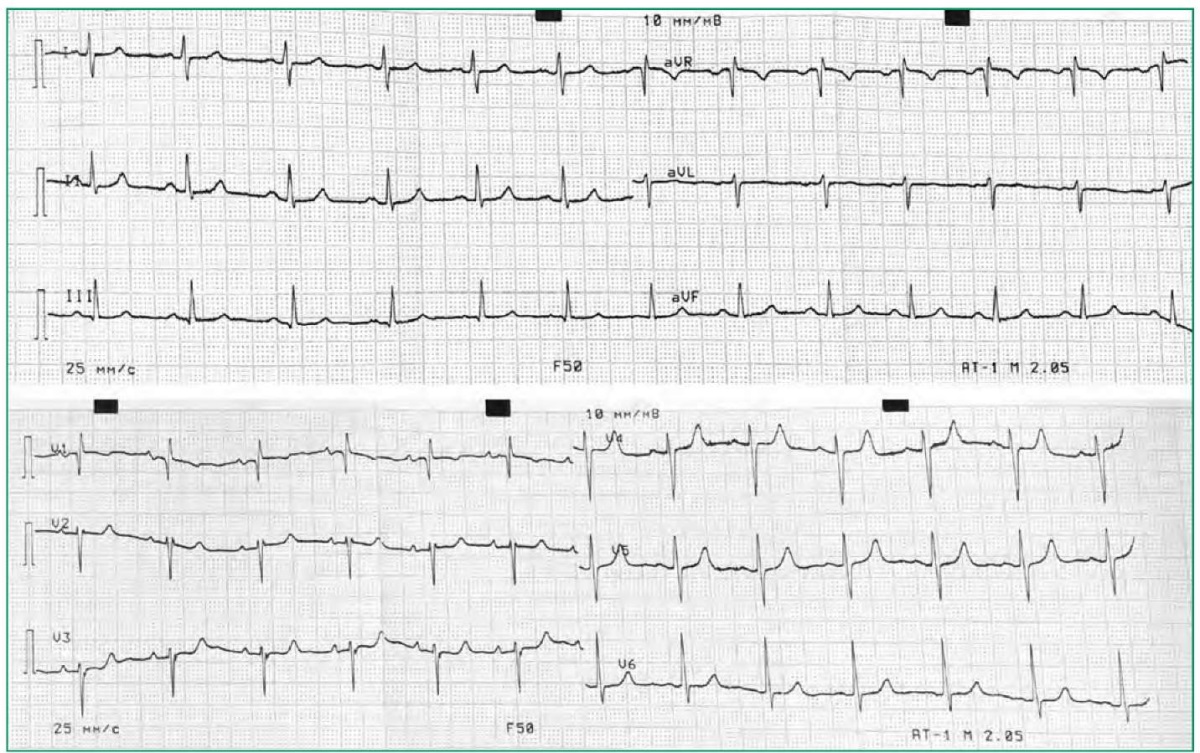
Figure 2. Electrocardiogram of patient T. (description in the text).
Рисунок 2. Электрокардиограмма пациентки Т. (описание в тексте).
При эхокардиографии (рис. 3) отмечалось умеренное расширение обоих предсердий (левое 75 мл, правое 78 мл) и правого желудочка (4,0 см) с гиперкинезом его свободной стенки, левый желудочек не расширен, его фракция выброса составила 65%. Присутствовала гипертрофия стенок левого желудочка до 12 мм с нарушением его диастолической функции (E/A 1,97) в рамках не в полной мере, контролируемой на протяжении многих лет гипертонической болезни. Обращали на себя внимание расширение ствола легочной артерии (2,8 см) и признаки значительной легочной гипертензии (систолическое давление в легочной артерии 69 мм рт.ст., диастолическое давление 19 мм рт.ст.). Отмечались трикуспидальная регургитация 2 степени (скорость регургитации 3,2 м/с), легочная регургитация 1-2 степени.
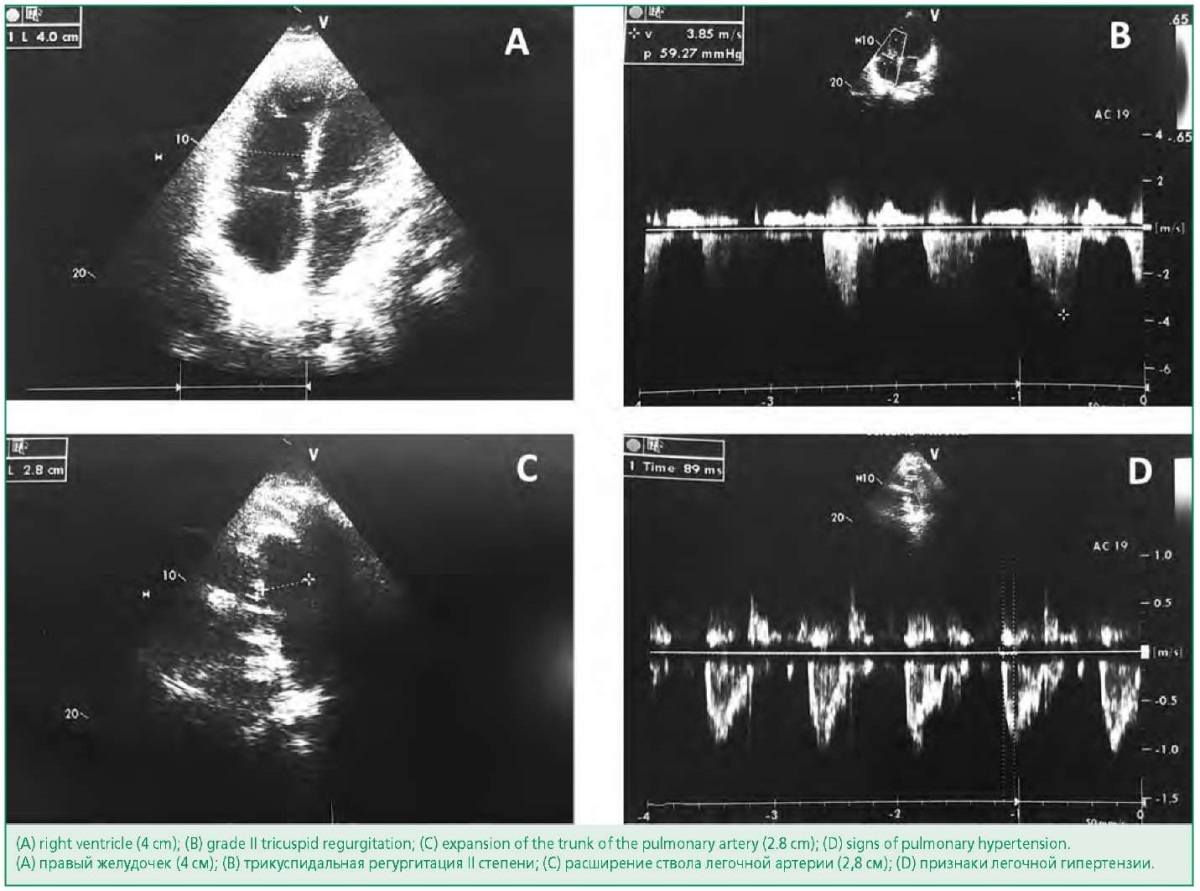
Figure 3. Echocardiography of patient T.
Рисунок 3. Эхокардиография пациентки Т.
При ультразвуковом исследовании брюшной полости визуализировалась увеличенная в сагиттальном размере печень, свободной жидкости в брюшной полости не выявлено. Для уточнения природы легочной гипертензии проведена мультиспиральная компьютерная томография легких (МСКТ) с внутривенным контрастированием (рис. 4), при которой данных за тромбоэмболию легочной артерии не получено, однако воздушность легочной ткани была диффузно неравномерно снижена за счет множественных зон повышения плотности по типу «матового стекла» (плотность легочной паренхимы до 660 едН). На этом фоне, больше в базальных отделах определялись множественные воздушные ловушки. Такая МСКТ-картина может быть обусловлена гиперсенситивным пневмонитом или десквамативной интерстициальной пневмонией. Проведена бодиплетизмография, при которой отмечены признаки рестриктивных вентиляционных нарушений легкой степени (форсированная жизненная емкость легких 70%), признаки нерезко выраженной гипервоздушности легочной ткани (остаточный объем / остаточная емкость легких = 0,52 при остаточном объеме 93%), легкое снижение диффузионной способности легких (DLcocor = 67%). Пациентка была проконсультирована торакальным хирургом, член-корр. РАН В.Д. Паршиным: риск торакоскопической биопсии легкого для морфологической верификации поражения легких представляется неоправданно высоким с учетом степени легочной гипертензии, риском кровотечения выше среднего (в связи с болезнью Рандю-Ослера-Вебера), процедура может выполняться только по жизненным показаниям. Принято решение о проведении пульс-терапии преднизолоном внутривенно 240 мг/сут в течение 3-х дней с последующим переходом на пероральный прием глюкокортикоидов. Контрольная МСКТ после курса терапии показала небольшую отрицательную динамику изменений в легких в виде повышения плотности легочной паренхимы в зонах «матового стекла».
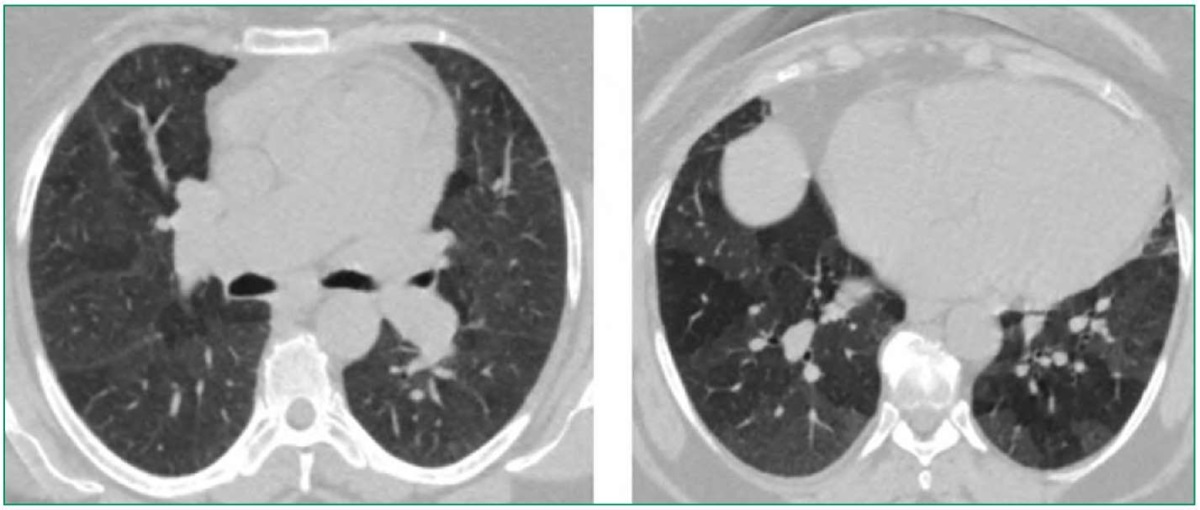
Figure 4. Multispiral computed tomography of the chest of patient T. (description in the text).
Рисунок 4. Мультиспиральная компьютерная томография органов грудной клетки пациентки Т. (описание в тексте).
Клинический диагноз был сформулирован как: Болезнь Рандю-Ослера-Вебера: геморрагический синдром с частыми носовыми кровотечениями. Гипертоническая болезнь II стадии, 3 степени повышения АД, высокого риска. Нарушения ритма сердца: наджелудочковая и желудочковая экстрасистолия, неустойчивая желудочковая тахикардия. Тяжелая легочная гипертензия. Дыхательная недостаточность II степени. Хроническое легочное сердце в фазе субкомпенсации. Хроническая сердечная недостаточность с сохранной фракцией выброса IIБ стадии, 3 функциональный класс по NYHA. Железодефицитная анемия легкой степени, длительно леченная препаратами железа. Ожирение 2 степени.
Проводился поиск возможной причины сердечной недостаточности. Несмотря на адекватную терапию хронической сердечной недостаточности и анемии, пациентка отмечала сохранение слабости, SpО2 оставалась сниженной. При эхокардиографии зарегистрированы высокая легочная гипертензия, перегрузка правых отделов сердца. С целью выявления причины легочной гипертензии выполнена МСКТ легких с внутривенным контрастированием. МСКТ-картина и данные бодиплетизмографии соответствовали интерстициальному характеру поражения легких, однако от морфологической верификации было решено воздержаться. С учетом отсутствия ответа на терапию глюкокортикоидами диагноз интерстициального заболевания легких был отвергнут. Нормальный уровень D-димера и данные МСКТ позволили исключить тромбоэмболию мелких ветвей легочной артерии. Пациентка была проконсультирована генетиком, проф. Е.В. Заклязьминской, взята кровь для проведения ДНК-диагностики в гене ENG. Кроме того, проконсультирована доцентом кафедры пульмонологии к.м.н. Н.А. Царевой – подтверждено предположение о прямой патогенетической связи легочной гипертензии и изменений по типу «матового стекла» с болезнью Рандю-Ослера-Вебера, рекомендованы анализы крови на антинуклеарный фактор и антитела к ДНК-топоизомеразе I (анти-Scl-70; для исключения редкой формы склеродермии) с последующим включением в программу лечения легочной гипертензии антагонистом эндотелиновых рецепторов мацитентаном.
В качестве терапии пациентка получала бисопролол 5 мг/сут, лозартан 100 мг/сут, амлодипин 5 мг/сут, спиронолактон 50 мг/сут, фуросемид 60-80 мг/сут внутривенно с переходом на пероральный прием фуросемида 120 мг/сут, омепразол 40 мг/сут, железа карбоксимальтозат 500 мг/сут №1, преднизолон внутривенно 240 мг/сут №3, метилпреднизолон 24 мг/сут – 2 дня после пульс-терапии с последующей постепенной отменой. В связи с дыхательной недостаточностью проводились ингаляции увлажненного кислорода. Во избежание подъемов АД, которые провоцируют носовые кровотечения, усилена антигипертензивная терапии (добавлен амлодипин). Прием фуросемида проводился под контролем суточного диуреза. В результате достигнут положительный диурез +400- 500 мл/сут. Постгеморрагическая анемия скорректирована внутривенной инфузией 500 мг железа карбоксимальтозата. Концентрация гемоглобина при выписке – 122 г/л. В результате проведенного лечения отмечена положительная динамика в виде значительного уменьшения одышки, регресса отеков нижних конечностей, АД и ЧСС установились на целевом уровне.
После выписки из стационара были сданы анализы крови на антинуклеарный фактор и антитела к ДНКтопоизомеразе I (анти-Scl-70), данных за наличие склеродермии получено не было, пациентке был назначен мацитентан, однако от его применения пациентка воздержалась в связи с тем, что анемия фигурировала в инструкции как один из частых побочных эффектов. Состояние пациентки оставалось стабильным до августа 2020 г., когда остро наросла одышка и бригадой скорой медицинской помощи больная была госпитализирована в отделение реанимации больницы по месту жительства, где, спустя несколько часов, умерла. Причина смерти при аутопсии расценена как астматический статус, бронхиальная астма.
В данном клиническом случае сочетается болезнь Рандю-Ослера-Вебера и тяжелая легочная гипертензия с интерстициальным поражением легких. Легочная артериальная гипертензия (ЛАГ) сама по себе также является достаточно редким заболеванием с частотой 15-25 случаев на миллион [6]. В основе развития первичной ЛАГ в 75% случаев лежат мутации в гене рецептора костного морфогенетического белка 9 типа BMPR2 [7], однако в остальных 25% мутации выявляются в иных генах, в том числе ACVLR1, ENG, SMAD4, и GDF2, ассоциированных с НГТ [8]. Именно с общей генетической основой, ведущей к нарушению микроангиогенеза, связано формирование перекрестного фенотипа, при котором сочетаются НГТ и ЛАГ [9]. В ряде случаев первым проявлением заболевания становится именно ЛАГ, и лишь в дальнейшем ставится диагноз НГТ [10][11]. В нашем случае наоборот пациентка длительно страдала НГТ, а первые проявления ЛАГ развились спустя 30 лет после дебюта болезни Рандю-Ослера-Вебера. Причем, появление признаков застоя по большому кругу кровообращения было сперва спровоцировано анемией, и в дальнейшем симптомы сердечной недостаточности нарастали на фоне новых эпизодов кровопотери, усугубляющих железодефицит. Генетический дефект, ставший причиной сочетания НГТ и ЛАГ у пациентки, пока не установлен. На данный момент, в связи с ограниченными финансовыми возможностями, проведен только анализ гена ENG, мутации в котором обнаружены не были, однако посмертно планируется оценить и другие три гена, ассоциированные с НГТ, а при отсутствии в них мутаций, выполнить полноэкзомное секвенирование.
Еще одна особенность представленного клинического случая – интерстициальное поражение легких. Эти изменения не являются, по всей видимости, ни самостоятельным интерстициальным заболеванием легких, поскольку не было получено ответа на пульстерапию глюкокортикостероидами, ни пневмонией, т.к. полностью отсутствовали аускультативные изменения, кашель и провоспалительные изменения в крови. Вероятно, изменения по типу «матового стекла» обусловлены перераспределением крови в рамках болезни Рандю-Ослера-Вебера в сочетании с высокой легочной гипертензией. В литературе описан лишь один случай сочетания НГТ, ЛАГ и интерстициального поражения легких: это женщина 65 лет с семейной формой НГТ с ранним дебютом; проявления сердечной недостаточности, как и в нашем случае, возникли довольно поздно, в 50 лет, а при МСКТ, выполненной в 65 лет для исключения артериовенозных мальформаций в легких, были выявлены изменения по типу «матового стекла» [12]. К сожалению, ДНК-диагностика этой пациентке не проводилась.
Лечение пациентов с НГТ, помимо профилактики и своевременной остановки кровотечений, не разработано. Есть лишь экспериментальные работы, демонстрирующие эффект от препаратов, способных ингибировать патологический ангиогенез, таких как бевацизумаб, ингибиторы тирозин-киназы, а также такролимус и сиролимус [2]. Данные относительно лечения легочной гипертензии у пациентов с НГТ ограничены, однако имеются данные, демонстрирующие хороший ответ на стандартное лечение ЛАГ [9]. Одной из перспективных молекул для лечения сочетания НГТ и ЛАГ считается регулятор транскрипции ID3, т.к. он патогенетически связан с каскадами, ведущими к развитию обоих заболеваний, однако это требует дальнейшего изучения [13][14].
Смерть пациентки, наиболее вероятно, наступила не от астматического статуса, т.к. больная никогда не страдала бронхиальной астмой, а от значительного повышения давления в системе легочной артерии, что могло привести к отеку легких. Возможно, проведение ЛАГ-специфической терапии (от которой пациентка воздержалась) могло бы отсрочить или даже предотвратить неблагоприятный исход.
Описанный нами клинический случай имеет ряд ограничений, связанных с техническими возможностями: не проводились катетеризация сердца, определение уровня натрийуретического пептида и полноэкзомное секвенирование. В связи с выраженной общей слабостью пациентки, обусловленной анемией, не был выполнен тест с шестиминутной ходьбой. Это затрудняло точную оценку класса ЛАГ и определение прогноза, однако последнее, с учетом летального исхода, на момент описания данного клинического случая утратило свою актуальность.
Болезнь Рандю-Ослера-Вебера, помимо сосудистых проявлений, может манифестировать высокой легочной гипертензией и интерстициальным поражением легких. Прогноз при НГТ в сочетании с ЛАГ определяется не только интенсивностью кровотечений, но и степенью выраженности легочной гипертензии, которую необходимо своевременно выявлять и лечить при помощи ЛАГ-специфической терапии. Интерстициальное поражение легких при болезни Рандю-Ослера-Вебера является проявлением основного заболевания и не требует отдельного лечения.
Отношения и Деятельность. Нет.
Relationships and Activities. None.
1. Kühnel T, Wirsching K, Wohlgemuth W, et al. Hereditary Hemorrhagic Telangiectasia. Otolaryngol Clin North Am. 2018;51(1):237-54. DOI:10.1016/j.otc.2017.09.017.
2. Robert F, Desroches-Castan A, Bailly S, et al. Future treatments for hereditary hemorrhagic telangiectasia. Orphanet J Rare Dis. 2020;15(1):1-10. DOI:10.1186/S13023-019-1281-4.
3. Govani FS, Shovlin CL. Hereditary haemorrhagic telangiectasia: a clinical and scientific review. Eur J Hum Genet. 2009;17(7):860-71. DOI:10.1038/EJHG.2009.35.
4. David L, Mallet C, Mazerbourg S, et al. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood. 2007;109(5):1953-61. DOI:10.1182/BLOOD-2006-07-034124.
5. Dupuis-Girod S, Bailly S, Plauchu H. Hereditary hemorrhagic telangiectasia: from molecular biology to patient care. J Thromb Haemost. 2010;8(7):1447-56. DOI:10.1111/J.1538-7836.2010.03860.X.
6. Galiè N, Humbert M, Vachiery JL, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016;37(1):67-119. DOI:10.1093/EURHEARTJ/EHV317.
7. Soubrier F, Chung WK, Machado R, et al. Genetics and genomics of pulmonary arterial hypertension. J Am Coll Cardiol. 2013;62(25 Suppl):D13-21. DOI:10.1016/J.JACC.2013.10.035.
8. Tillet E, Bailly S. Emerging roles of BMP9 and BMP10 in hereditary hemorrhagic telangiectasia. Front Genet. 2015;5:456. DOI:10.3389/fgene.2014.00456.
9. Vorselaars VMM, Hosman AE, Westermann CJJ, et al. Pulmonary Arterial Hypertension and Hereditary Haemorrhagic Telangiectasia. Int J Mol Sci. 2018;19(10):3203. DOI:10.3390/IJMS19103203.
10. Ishiwata T, Terada J, Tanabe N, et al. Pulmonary arterial hypertension as the first manifestation in a patient with hereditary hemorrhagic telangiectasia. Intern Med. 2014;53(20):2359-63. DOI:10.2169/internalmedicine.53.2850.
11. Yokokawa T, Sugimoto K, Kimishima Y, et al. Pulmonary Hypertension and Hereditary Hemorrhagic Telangiectasia Related to an ACVRL1 Mutation. Intern Med. 2020;59(2):221-7. DOI:10.2169/INTERNALMEDICINE.3625-19.
12. Jain D, Viswanathan S, Ramasamy C. Hereditary Hemorrhagic Telangiectasia with Unusual Associations. Cureus. 2015;7(6):1-4. DOI:10.7759/CUREUS.278.
13. Avecilla V. Effect of Transcriptional Regulator ID3 on Pulmonary Arterial Hypertension and Hereditary Hemorrhagic Telangiectasia. Int J Vasc Med. 2019;2019:2123906. DOI:10.1155/2019/2123906.
14. Kim JH, Peacock MR, George SC, et al. BMP9 induces EphrinB2 expression in endothelial cells through an Alk1-BMPRII/ActRII-ID1/ID3-dependent pathway: implications for hereditary hemorrhagic telangiectasia type II. Angiogenesis. 2012;15(3):497-509. DOI:10.1007/S10456-012-9277-X.
Лутохина Юлия Александровна,
Москва
Благова Ольга Владимировна,
Москва
Савина Полина Олеговна,
Москва
Заклязьминская Елена Валерьевна,
Москва
Лутохина Ю.А., Благова О.В., Савина П.О., Заклязьминская Е.В. Болезнь Рандю-Ослера-Вебера с высокой легочной гипертензией и интерстициальным поражением легких. Рациональная Фармакотерапия в Кардиологии. 2023;19(2):179-185. https://doi.org/10.20996/1819-6446-2023-03-06
Lutokhina Yu.A., Blagova O.V., Savina P.O., Zaklyazminskaya E.V. Rendu-Osler-Weber Disease with High Pulmonary Hypertension and Interstitial Lung Disease. Rational Pharmacotherapy in Cardiology. 2023;19(2):179-185. (In Russ.) https://doi.org/10.20996/1819-6446-2023-03-06

.jpg)
Главный редактор
 Драпкина О. М.
Драпкина О. М.
101990, Москва, Петроверигский пер., 10,
Тел.: +7 (499) 553 68 10
е-mail: otsec.rfc@mail.ru







































