


Содержание
Перейти к:

 О. В. Благова,
Е. В. Павленко,
Н. В. Вариончик,
В. П. Седов,
Н. В. Гагарина,
Е. А. Мершина,
М. Е. Поляк,
Е. В. Заклязьминская,
А. В. Недоступ
О. В. Благова,
Е. В. Павленко,
Н. В. Вариончик,
В. П. Седов,
Н. В. Гагарина,
Е. А. Мершина,
М. Е. Поляк,
Е. В. Заклязьминская,
А. В. Недоступ https://doi.org/10.20996/1819-6446-2022-02-01
Перейти к:
Цель. Изучить место некомпактного миокарда (НКМ) в структуре дилатационной кардиомиопатии (ДКМП), его клинические особенности и влияние на прогноз по сравнению с другими формами синдрома ДКМП.
Материалы и методы. В регистр НКМ включены 125 больных, средний возраст 46,4±15,1 лет, 74 мужчины и 51 женщина, медиана срока наблюдения 14 [4,0; 41,0] мес. В регистр ДКМП включено 365 пациентов, средний возраст 46,4±15,1 лет, 253 мужчины и 112 женщин, медиана срока наблюдения 14 [5; 43,75] мес. Обследование включало электрокардиографию (ЭКГ), Холтеровское мониторирование ЭКГ, эхокардиографию, оценку уровня антикардиальных антител в крови, а также мультиспиральную компьютерную и магнитно-резонансную томографию сердца, ДНК-диагностику (в генах МYH7, MYBPC3, TPM1, TNNI3, TNNT2, ACTC1, TAZ, ZASP (LDB3), MYL2, MYL3, DES, LMNA, EMD, TTR), коронароангиографию, эндомиокардиальную биопсию правого желудочка.
Результаты. Доля пациентов с фенотипом ДКМП в регистре НКМ составила 40% (n=49), еще 11% (n=15) имели НКМ, диагностированный одновременно с острым/подострым миокардитом. Летальность в этих подгруппах составила 12,2% и 33,3% соответственно, и была значительно выше, чем при бессимптомном, ишемическом и аритмическом вариантах НКМ. В регистре ДКМП доля больных с НКМ составила 21% (n=78), еще у 18% (n=64) была выявлена повышенная трабекулярность левого желудочка (ЛЖ). Больные ДКМП с НКМ и без НКМ не различались по исходным эхокардиографическим параметрам, классу сердечной недостаточности и кардиотропной терапии. Патогенные мутации выявлены у 14% больных ДКМП с НКМ и всего у 3% остальных пациентов с ДКМП (p<0,001). Только у пациентов без НКМ наличие мутаций существенно влияло на летальность. У пациентов с НКМ по сравнению с остальными больными отмечалось значимо меньшее увеличение фракции выброса ЛЖ в ранние и поздние сроки (с 31,0±10,2 до 34,8±11,0 и 37,1±10,9% [р<0,05] и с 31,8±9,7 до 38,8±11,3 и 42,3±12,4% [р<0,01] соответственно), бо́льшая частота желудочковых экстрасистол (1568 [105;7000] против 543,5 [77,75; 3194], p<0,05), оправданных шоков дефибриллятора и внезапных смертей (17,9 против 5,9%, p<0,001), внутрисердечного тромбоза (21,8 против 13,5%, p=0,069), несмотря на более высокую частоту применения антикоагулянтов (73,1 против 57,4%, p<0,05). Существенной разницы в смертности (19,2% против 18,5%) и частоте трансплантации (7,7% против 3,8%) между пациентами с НКМ и без НКМ не было. Случаев регрессии НКМ не отмечено.
Заключение. НКМ является самостоятельной формой синдрома ДКМП, которая характеризуется более высокой частотой патогенных мутаций, аритмических событий, худшей реакцией на кардиотропную терапию, более высокой частотой внутрисердечного тромбоза. Отсутствие различий в смертности можно объяснить более высокой частотой профилактических вмешательств у данной категории пациентов с ДКМП (назначение антикоагулянтов, имплантация дефибриллятора, трансплантация сердца).
Благова О.В., Павленко Е.В., Вариончик Н.В., Седов В.П., Гагарина Н.В., Мершина Е.А., Поляк М.Е., Заклязьминская Е.В., Недоступ А.В. Некомпактный миокард с дилатационным фенотипом: проявления, лечение и исходы в сравнении другими формами синдрома дилатационной кардиомиопатии. Рациональная Фармакотерапия в Кардиологии. 2022;18(1):27-35. https://doi.org/10.20996/1819-6446-2022-02-01
Blagova O.V., Pavlenko E.V., Varionchik N.V., Sedov V.P., Gagarina N.V., Mershina E.A., Polyak M.E., Zaklyazminskaya E.V., Nedostup A.V. Noncompact Myocardium with Dilated Phenotype: Manifestations, Treatment and Outcomes in Comparison with Other Forms of Dilated Cardiomyopathy Syndrome. Rational Pharmacotherapy in Cardiology. 2022;18(1):27-35. https://doi.org/10.20996/1819-6446-2022-02-01
Некомпактный миокард (НКМ) остается одним из наименее изученных вариантов кардиомиопатий (КМП). В европейской классификации КМП он отнесен к неклассифицируемым КМП [1], отсутствуют рекомендации по его диагностике и лечению, специально спланированные исследования НКМ, симпозиумы на конгрессах в Европе.
Остается нерешенным вопрос о природе НКМ. Не вызывает сомнений генетическая обусловленность феномена НКМ [2], однако слишком большое количество и разнородность генов, мутации в которых уже описаны у пациентов, высокая частота сочетаний НКМ с другими КМП (гипертрофической, рестриктивной) размывают границы и заставляют сомневаться в нозологической обособленности НКМ. Высказывается мнение о возможной вторичности НКМ – вследствие значительной дилатации камер сердца, его перегрузок (спорт, беременность), некоторых заболеваний (серповидноклеточная анемия) [3]. В целом вопрос формулируется следующим образом: НКМ – признак, феномен или болезнь [4]?
В то же время число генов, потенциально ответственных за развитие дилатационной кардиомиопатии (ДКМП), еще больше, однако это не заставляет сомневаться в существовании первичной (истинной) ДКМП, в отличие от иных вариантов синдрома ДКМП [5]. Вероятно, в случае НКМ также можно использовать термин «синдром», хотя само наличие НКМ, на наш взгляд, склоняет диагноз в пользу генетически детерминированной КМП. Наибольшие вопросы вызывает именно НКМ с дилатационным фенотипом. Если при других формах КМП говорят о сочетании с НКМ, то при наличии картины ДКМП чаще расценивают ее как наиболее типичный фенотип НКМ (так называемая некомпактная КМП).
Задачей настоящего исследования не было окончательно ответить на вопрос о природе НКМ, но для практической кардиологии важны менее общие вопросы: 1) есть ли отличия у пациентов с НКМ и дилатационным фенотипом от прочих больных с синдромом ДКМП; 2) требуют ли эти отличия особых подходов к лечению больных с НКМ и фенотипом ДКМП; 3) означает ли наличие НКМ худший прогноз в сопоставлении с другими вариантами синдрома ДКМП.
Цель нашего исследования – изучение места НКМ в структуре ДКМП, его клинических особенностей и влияния на прогноз по сравнению с другими формами синдрома ДКМП.
В регистр НКМ включены 125 больных (средний возраст 46,4±15,1 лет; 74 мужчины и 51 женщина; медиана срока наблюдения 14 [ 4,0; 41,0] мес).
Критерии включения: возраст ≥16 лет; наличие визуальных критериев НКМ: двуслойный миокард левого желудочка (ЛЖ) с соотношением некомпактного к компактному от 2:1 при эхокардиографии (ЭхоКГ) или от 2,3 к 1 при мультиспиральной компьютерной томографии/магнитно-резонансной томографии; синхронное движение некомпактного и компактного слоев; выявление более 3 трабекул в ЛЖ и наличие межтрабекулярного кровотока в конце диастолы.
В регистр ДКМП включены 365 больных (средний возраст 46,4±15,1 лет; 253 мужчины и 112 женщин; медиана срока наблюдения 14 [ 5; 43,75] мес).
Критерии включения: возраст ≥16 лет, дилатация ЛЖ (конечный диастолический размер >5,5 см) и его систолическая дисфункция (фракция выброса [ФВ] <50%).
Критерии невключения: инфаркт миокарда, острый коронарный синдром вследствие коронарного атеросклероза, инфекционный эндокардит давностью менее 6 мес, приобретенные пороки сердца, тиреотоксическое, гипертоническое сердце (гипертрофия миокарда ЛЖ >14 мм), верифицированный амилоидоз, болезни накопления, саркоидоз, диффузные болезни соединительной ткани, системные васкулиты, операции на сердце давностью менее 2 мес, отказ пациента от участия.
Исследование одобрено межвузовским этическим комитетом. Все пациенты подписали добровольное информированное согласие на дополнительные обследования.
Методы исследования включали электрокардиографию, Холтеровское мониторирование электрокардиограммы, эхокардиографию (ЭхоКГ), оценку уровня антикардиальных антител в крови непрямым методом иммунофлюоресценции, а также мультиспиральную компьютерную томографию, магнитно-резонансную томографию сердца, ДНК-диагностику [в генах МYH7, MYBPC3, TPM1, TNNI3, TNNT2, ACTC1, TAZ, ZASP (LDB3), MYL2, MYL3, DES, LMNA, EMD, TTR], коронароангиографию, эндомиокардиальную биопсию правого желудочка. Цель расширенного обследования – выявление критериев исключения, уточнение нозологической природы синдрома ДКМП (верификация диагноза миокардита). Клинические варианты (маски) НКМ определялись в соответствии с описанными ранее критериями [6].
Поиск мутаций был выполнен с использованием таргетной панели генов, с двумя пулами праймеров, дизайн которых был выполнен с помощью автоматического он-лайн ресурса Ion AmpliSeq Designer® (Thermo Fisher Scientific, США). Приготовление библиотек было выполнено с использованием Ion AmpliSeq™ Library Kit 2.0 в соответствии с протоколом производителя (Thermo Fisher Scientific). Секвенирование было выполнено на чипах Ion 314™ и Ion 316™ с использованием высокопроизводительного полупроводникового секвенатора Ion PGM™ System. Данные, полученные с Ion PGM™ System, были обработаны при помощи плагинов CoverageAnalysis и VariantCaller, входящих в лицензированное программное обеспечение Torrent Suite Software 5.6.0 и Ion Reporter Software (Thermo Fisher Scientific). Визуализация NGS прочтений проводилась с использованием Integrative Genomic Viewer, выравнивание осуществлялось на референсный геном по версии hg19. Частоты выявленных генетических вариантов, а также биоинформатический анализ in silico выполнялся с использованием внутреннего пайплайна, с использованием критериев ACMG (2015) [7], а также баз данных Genome Aggregation Database, ClinVar, HGMD, он-лайн ресурсов VarSome, PolyPhen 2.0, HSF, CardioDB.
Первичные конечные точки включали летальность, частоту внезапной сердечной смерти (ВСС), трансплантации сердца и показатель «смерть+трансплантация», вторичные – показатель «оправданные срабатывания дефибрилляторов+ВСС», частоту внутрисердечного тромбоза, эмболических событий, динамику ФВ.
Лечение больных с синдромом ДКМП (как с НКМ, так и без него) проводилось в соответствии с европейскими и российскими рекомендациями и включало оптимальную медикаментозную терапию хронической сердечной недостаточности (ХСН), имплантацию кардиовертеров-дефибрилляторов и ресинхронизирующих устройств с функцией дефибриллятора, а также проведение базисной терапии миокардита в случае его верификации. О назначении непрямых антикоагулянтов будет сказано отдельно.
Статистическая обработка материала проводилась с помощью программы SPSS Statistics 22.0 (IBM, США). Количественные признаки представлены как среднее (M) и стандартное отклонение (δ), либо в виде медианы (Me) и интерквартильного диапазона [ 25%; 75%]. Нормальность распределения оценивалась с помощью теста Колмогорова-Смирнова, статистическую значимость различий – с помощью критериев Стьюдента, Манна-Уитни, Уилкоксона. Для оценки выживаемости проводилось построение кривых Каплана-Майера. Различия считались значимыми при р<0,05.
Доля больных с НКМ и фенотипом ДКМП в регистре больных с НКМ составила 40% (n=49), еще 11% (n=15) имели синдром ДКМП, диагностированный одновременно с острым/подострым миокардитом (рис. 1). Летальность в этих подгруппах составила соответственно 12,2% и 33,3%, и была выше, чем при клинических вариантах НКМ без значимой дилатации камер – бессимптомном, ишемическом и аритмическом вариантах ДКМП (табл. 1).
Table 1. Clinical manifestations of non-compacted myocardium with dilated cardiomyopathy phenotype, including combination with acute/subacute myocarditis, in comparison with other non-compacted myocardium variants
Таблица 1. Клинические проявления НКМ с фенотипом ДКМП, включая сочетание с острым/подострым миокардитом, в сопоставлении с другими вариантами НКМ
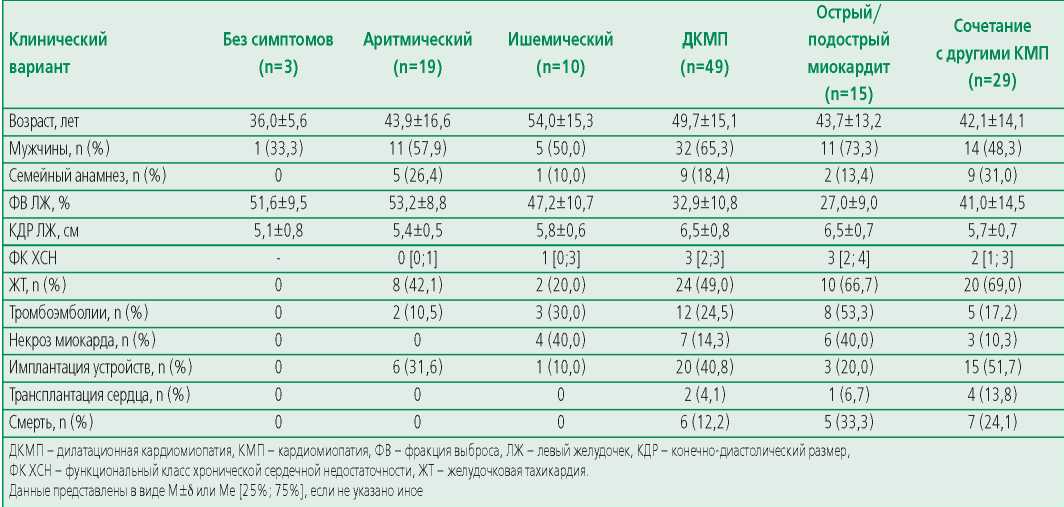
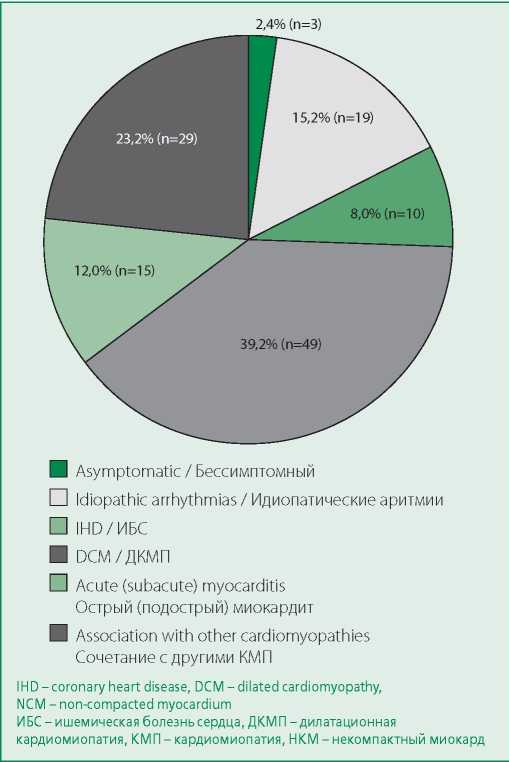
Figure 1. Frequency of different clinical variants in registry of patients with NСM (n=125)
Рисунок 1. Частота различных клинических вариантов в регистре больных НКМ (n=125)
При сопоставлении по показателю «смерть+трансплантация» прогноз пациентов с фенотипом ДКМП оказался существенно хуже, чем при вариантах без значимой ХСН, особенно – при сочетании с острым/подострым миокардитом (рис. 2). У части больных с «хронической» ДКМП (без острого дебюта давностью менее полугода) также встречался миокардит, однако он не определял остроту и тяжесть декомпенсации. Наихудший прогноз отмечен при сочетании с другими КМП (рестриктивной, гипертрофической, аритмогенной правожелудочковой), однако дилатация камер и систолическая дисфункция возникали лишь у некоторых больных на поздних стадиях.
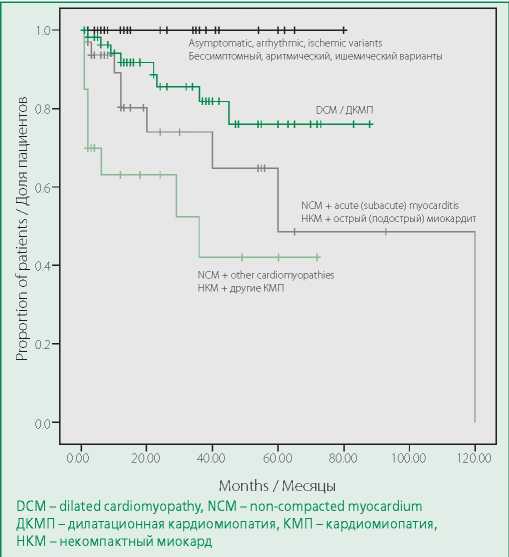
Figure 2. Kaplan-Meier curves for the “death+transplant” rate in the study groups
Рисунок 2. Кривые Каплана-Майера для показателя «смерть+трансплантация» в исследуемых группах
Частота НКМ в регистре больных с синдромом ДКМП представлена на рис. 3. Общая доля пациентов с НКМ в регистре больных ДКМП составила 21% (n=78). Еще у 64 пациентов с ДКМП была выявлена повышенная трабекулярность ЛЖ – соотношение толщины некомпактного и компактного слоев от 1 до 2. Пациенты с НКМ не отличались от других больных с синдромом ДКМП по основным демографическим и эхокардиографическим показателям, выраженности ХСН, объему кардиотропной терапии (табл. 2). Дальнейшее сравнение касалось более специфичных для НКМ параметров – генетической основы, аритмических и тромбоэмболических событий, а также исходов.
Table 2. Baseline clinical and demographic characteristics of patients with dilated cardiomyopathy syndrome with or without non-compacted myocardium
Таблица 2. Исходные клинико-демографические характеристики больных с синдромом ДКМП с или без НКМ
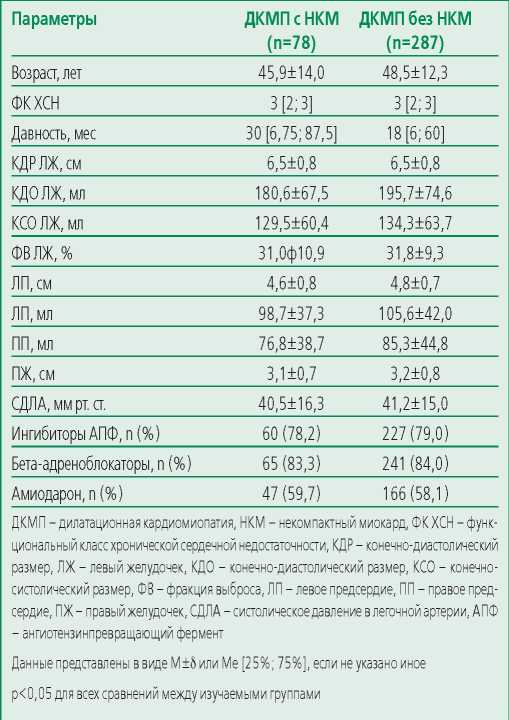
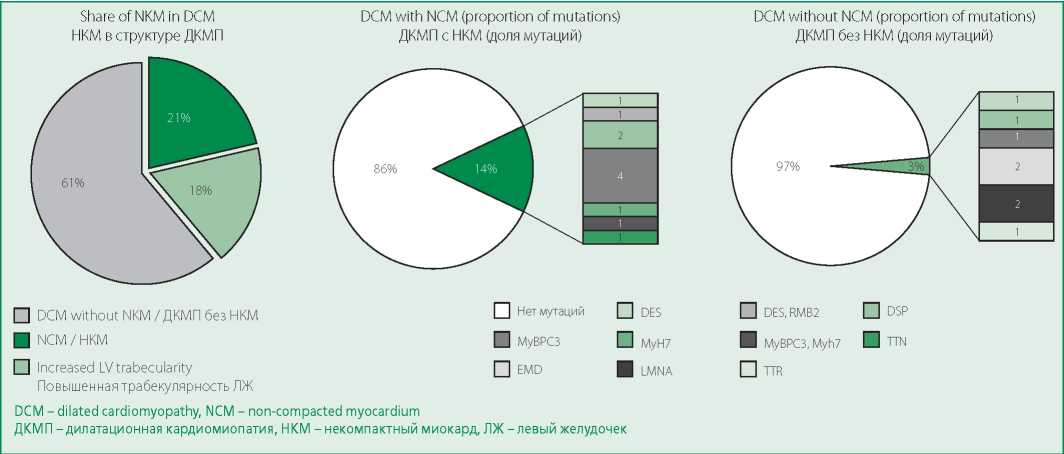
Figure 3. Proportion of noncompact myocardium in the structure of DCM registry and features of its genetic nature in comparison with other variants of DCM
Рисунок 3. Доля некомпактного миокарда в структуре регистра ДКМП и особенности его генетической природы в сравнении с другими вариантами ДКМП
Частота выявления мутаций у больных с НКМ оказалась значимо выше, чем у пациентов с ДКМП без НКМ, и составила 14% и 3% соответственно (p<0,001; см. рис. 3). При этом у больных с НКМ преобладали мутации в генах саркомерных белков (чаще всего – в гене MyBPC3), в то время как у пациентов без НКМ чаще всего удавалось выявить генетическую природу при наличии скелетной миопатии (мутации в генах DES, LMNA, EMD). При оценке прогностической значимости выявленных мутаций отмечено их значимое отрицательное влияние на прогноз (частоту достижения показателя «смерть+трансплантация») у пациентов с ДКМП без НКМ, в то время как у больных с НКМ прогноз не менялся в зависимости от обнаружения генетической природы болезни (рис. 4).
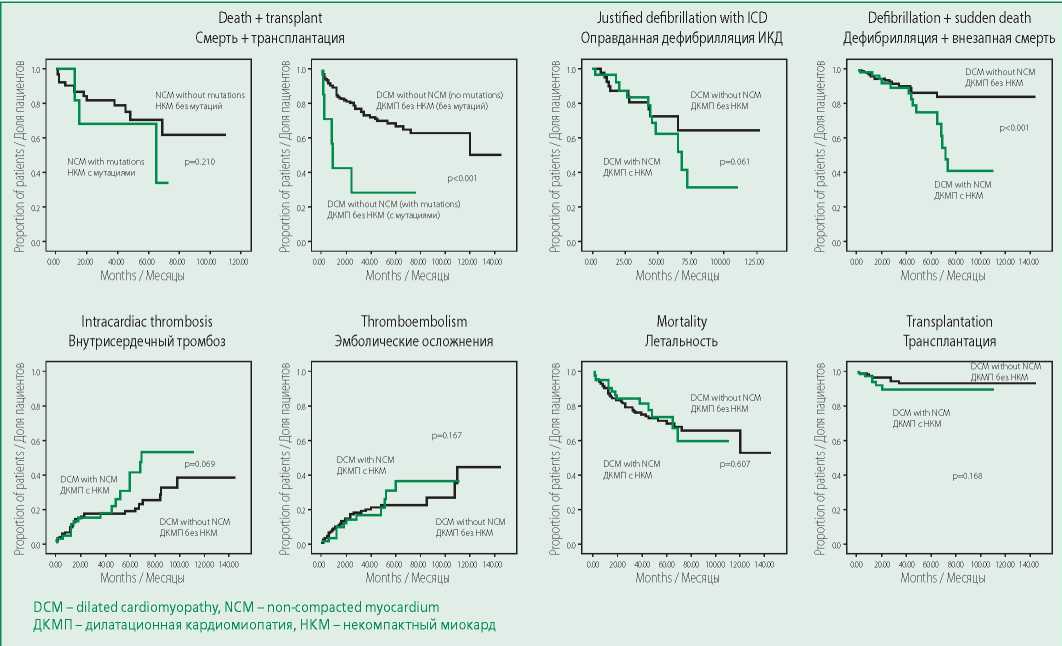
Figure 4. Frequency of complications and unfavorable outcomes in patients with noncompaction myocardium compared to other patients with DCM
Рисунок 4. Частота осложнений и неблагоприятных исходов у больных с некомпактным миокардом в сравнении с остальными пациентами с ДКМП
Изучено влияние некомпактного миокарда на результаты комплексного лечения ХСН (прирост сократительной функции ЛЖ). При отсутствии значимых различий по исходной ФВ ЛЖ между группами ДКМП без НКМ и ДКМП с НКМ (31,8±9,7 против 31,0±10,2%; p>0,05) отмечено значимо худшее ее возрастание как в первые 2-4 мес лечения (непосредственный ответ на лечение: 38,8±11,3% против 34,8±11,0%; p<0,05), так и к концу срока наблюдения (42,3±12,4% против 37,1±10,9% соответственно; p<0,01). При этом между пациентами с НКМ и без него не было значимых различий по частоте миокардита (по данным морфологического и комплексного обследования), что позволяет говорить о самостоятельном влиянии наличия НКМ на результаты лечения. Ни в одном случае не отмечено регресса НКМ в результате улучшения сократимости и сокращения размеров ЛЖ.
К типичным как для НКМ, так и для синдрома ДКМП нарушениям ритма относятся фибрилляция предсердий (ФП), желудочковая экстрасистолия (ЖЭ), устойчивая и неустойчивая тахикардия (ЖТ), а также ВСС вследствие фибрилляции желудочков. Медиана количества ЖЭ у пациентов с НКМ оказалась значимо выше, чем у больных без НКМ (1568 [ 105; 7000] против 544 [ 77; 3216] ЖЭ/сут; p<0,001). Неустойчивая ЖТ регистрировалась у 54% и 46% больных, устойчивая ЖТ – у 10% и 5%, блокада левой ножки пучка Гиса – у 30% и 26% (p>0,05 для всех).
С учетом большей частоты ЖЭ/ЖТ и меньшего прироста ФВ ЛЖ у больных с НКМ чаще принималось решение об имплантации дефибрилляторов. Устройства (кардиовертер-дефибриллятор и ресинхронизирующее устройство с функцией дефибриллятора) были имплантированы 32 больным с НКМ (41%) и 60 больным с иными вариантами ДКМП (21%), различия значимы (p<0,01). Дальнейший анализ показал целесообразность такого решения. Частота оправданных срабатываний дефибрилляторов была существенно выше у пациентов с НКМ в сравнении с больными без НКМ (34 и 17%, p=0,061), частота показателя «оправданные шоки+ВСС» была значимо выше у больных с НКМ (19% и 6%; p<0,001).
ФП несколько чаще встречалась у больных с ДКМП без НКМ, чем у пациентов с НКМ (58% и 67% соответственно). Тем не менее, непрямые антикоагулянты чаще назначались больным с НКМ (73% против 56%, p<0,05), поскольку в этом случае принималась во внимание также низкая ФВ ЛЖ, независимо от наличия ФП.
Кроме того, у больных с НКМ чаще выявлялся внутрисердечный тромбоз (28% против 13%, p=0,069), который тоже становился основанием для назначения антикоагулянтов. В результате значимых различий по частоте эмболических событий у больных с НКМ и без него выявлено не было (17 и 14% соответственно). В обеих группах наиболее частым эмболическим осложнением было острое нарушение мозгового кровообращения.
Проведена оценка частоты достижения конечных точек у больных с ДКМП и НКМ (срок наблюдения 22 [ 12; 47,25] мес) и больных с ДКМП без признаков НКМ (срок наблюдения 14 [ 5; 42,25] мес). Значимых различий не выявлено (см. рис. 4): частота летальных исходов составила 20,5% и 18,5%, трансплантация сердца выполнена 6 и 11 больным (7,7% и 3,8%), частота достижения конечной точки «смерть+трансплантация» составила 24,4% и 20,9%. Наиболее частой причиной смерти в обеих группах была терминальная ХСН.
Фенотип ДКМП хорошо известен как одно из самых типичных проявлений НКМ. Существует точка зрения, что только этот фенотип и является проявлением истинной «некомпактной кардиомиопатии». Вместе с тем наиболее крупные регистры НКМ как у детей, так и у взрослых включают этот фенотип наряду с другими – бессимптомным, гипертрофическим, рестриктивным и неопределенным [8][9]. Towbin J.A. и соавт. выделяют также смешанный (гипертрофический и дилатационный) фенотип [10].
Наши данные подтверждают разнородность фенотипических проявлений некомпактной КМП и позволяют проводить сравнительный анализ дилатационного и прочих фенотипов. Результаты этого анализа показали худший прогноз у больных с НКМ и фенотипом ДКМП в сравнении с бессимптомным, аритмическим и ишемическим вариантами (сочетание с подострым миокардитом, а также с иными формами КМП имело еще более серьезный прогноз). Эти данные полностью согласуются с другими исследованиями, в которых количественно преобладал дилатационный фенотип ДКМП (до 56% всех случаев НКМ [11]), и он имел даже более тяжелое течение, чем гипертрофический [8], что вполне ожидаемо.
Однако основной целью настоящего исследования было уточнить место НКМ среди всех пациентов с синдромом ДКМП. Аналогичный анализ у детей показал отсутствие различий по частоте смертей и трансплантаций у больных с НКМ и другими формами ДКМП [8], но сравнение по частоте жизнеугрожающих проявлений и видам лечения не проводилось. Более детальный анализ исходов у больных НКМ с дилатационным фенотипом показал, что лишь у 17% больных наступало улучшение (снижение класса ХСН), еще у 33% больных состояние оставалось стабильным, в то время как у второй половины больных заболевание протекало неблагоприятно – у 33% нарастал класс ХСН, 17% больных умерли [12].
Интересны данные, полученные нами в отношении прогностической значимости генетически верифицированных форм. Больные с НКМ, у которых к настоящему времени удалось выявить патогенные мутации, имели несколько худшую выживаемость, но все же значимо не отличались от больных, у которых мутации пока не обнаружены (ДНК-диагностика не завершена). Это можно считать косвенным свидетельством того, что и у оставшейся части больных НКМ имеет генетически детерминированную природу и протекает сходным образом. Частота выявления мутаций у больных с НКМ варьирует в диапазоне 20-30%, преобладание саркомерных мутаций подтверждено в большом недавнем метаанализе, включавшем 561 пациента с НКМ из 172 исследований [11]. Показана также связь мутаций в гене MyBPC3 (преобладавших у наших больных) с дебютом заболевания в среднем возрасте, систолической дисфункцией и неблагоприятными событиями в 31% случаев [13]. Однако ранее влияние наличия мутации на развитие ХСН и аритмий отмечено не было [14].
С другой стороны, в когорте наших больных с ДКМП без НКМ обнаружение патогенной мутации оказалось фактором негативного прогноза: мутации выявлялись значимо реже, чем при НКМ, и приводили к статистически значимому возрастание летальности. Объяснение этому факту видится в том, что доля истинной (первичной, генетически детерминированной) ДКМП в общей структуре синдрома ДКМП действительно мала – среди пациентов без выявленных мутаций такие больные тоже наверняка есть, но не они определяют прогноз.
Частота миокардита была достаточно высокой как у больных с НКМ, так и без него – но если при наличии НКМ миокардит лишь заметно утяжелял течение первичной КМП (и его успешное лечение все еще не обеспечивало хороших исходов), то у больных с прочими формами ДКМП миокардит часто был ведущей причиной декомпенсации, его лечение давало более заметные результаты. Именно этим в первую очередь можно объяснить выявленные нами значимые различия по выраженности положительного ответа на комплексную терапию, о которой судили по степени прироста ФВ (при отсутствии исходных различий). У больных с НКМ как непосредственный ответ на лечение, так и отдаленные его результаты оказались существенно хуже. Наличие НКМ сдерживало развитие положительных эффектов в т.ч. стандартной терапии ХСН, широко применявшейся у всех пациентов с ДКМП.
Установлены еще два клинических проявления, по которым больные с НКМ оказались тяжелее прочих больных с ДКМП – желудочковые аритмии и внутрисердечные тромбозы. При наличии НКМ оказались значимо чаще не только ЖЭ, но и конечная точка «оправданные срабатывания дефибрилляторов +ВСС». Различия по частоте тромбоза были незначимы, что подтверждает наличие специфических механизмов тромбообразования при наличии некомпактного слоя. Отметим, что оба феномена трудно было бы объяснить в рамках вторичного НКМ, обычного для тяжелой декомпенсации. В крупнейшем на сегодня метаанализе, включившем 2271 пациента с НКМ, устойчивая или неустойчивая ЖТ выявлена у 17%, тромбоэмболические события – у 9%, оправданные шоки дефибрилляторов – у 15%, т.е. несколько реже, чем в нашем исследовании [15].
Установленное при этом отсутствие значимых различий по частоте эмболических событий между нашими пациентами с НКМ и без него, несомненно, объясняется более частым назначением непрямых антикоагулянтов при НКМ. Показанием к такому лечению явились не только ФП и выявленный тромбоз, но и снижение ФВ ЛЖ менее 40%, как это рекомендуется в настоящее время [16][17]. Такой подход полностью себя оправдал. Кроме того, у пациентов с ДКМП без НКМ несколько чаще встречалась ФП, что могло повлиять на частоту эмболических событий в данной подгруппе и нивелировать различия.
Наконец, проведен анализ частоты достижения первичных конечных точек у больных с ДКМП и НКМ и без него. Установленное нами отсутствие значимых различий по общей летальности и частоте трансплантаций, тем не менее, не может рассматриваться как доказательство того, что наличие НКМ никак не влияет на прогноз у пациентов с ДКМП, как это предполагают некоторые авторы, проводившие сопоставление на основании магнитно-резонансной томографии [18].
В других работах такие различия все же были получены [19]. Нивелирование различий представляется нам закономерным (и желаемым) результатом активного воздействия на специфичные для НКМ механизмы танатогенеза – более частых имплантаций дефибрилляторов (при равных исходных ЭхоКГ-параметрах) и назначения антикоагулянтов. Третий механизм (недостаточный ответ на терапию при наличии НКМ) пока не подлежит коррекции (за исключением более раннего принятия решения о трансплантации сердца), однако и при иных вариантах ДКМП встречалась категория больных (и с миокардитом, и без него), плохо отвечавших на лечение, что требует дальнейшего изучения.
Ответим теперь на прикладные вопросы, которые были поставлены в начале этой работы. Отличия у пациентов с НКМ и дилатационным фенотипом от прочих больных с синдромом ДКМП (как генетической, так и негенетической и смешанной природы), безусловно, есть, причем именно эти отличия усугубляют риск неблагоприятных исходов и требуют особых подходов к лечению больных с НКМ и фенотипом ДКМП. Такие подходы разрабатываются, их активное применение в значительной мере позволяют позитивно влиять на прогноз больных с НКМ и сглаживать различия с иными вариантами ДКМП.
В заключение коснемся и фундаментального вопроса о сущности НКМ. Полученные данные позволяют утверждать, что феномен НКМ у больных с синдромом ДКМП в большинстве случаев является не вторичным следствием выраженной декомпенсации, но самостоятельным нозологическим вариантом. Это подтверждают следующие факты:
1. Феномен НКМ выявлялся и в отсутствие декомпенсации (и не объяснялся беременностью, занятиями спортом и пр.).
2. Пациенты с синдромом ДКМП иной этиологии, в т.ч. с тяжелым миокардитом и первичными формами имели не меньшую степень декомпенсации (не отличались ни по одному ЭхоКГ-параметру), однако НКМ у них не визуализировался.
3. Больные с дилатационным фенотипом НКМ имели специфические отличия клинической картины от прочих больных с ДКМП.
4. Регресс систолической дисфункции и сокращение размеров ЛЖ ни разу не сопровождались исчезновением феномена НКМ.
Ограничения исследования. По объективным причинам не у всех больных, включенных в исследование, завершена ДНК-диагностика, объем ее был различен у разных больных в зависимости от клинического фенотипа. Различались также сроки наблюдения за пациентами, однако в обеих исследуемых группах они были достаточными.
Наличие НКМ не приводит к исходно более выраженной дисфункции миокарда в сравнении с прочими больными с синдромом ДКМП, но сопровождается значимо худшим приростом ФВ в процессе лечения (в ранние и поздние сроки). Патогенные мутации (преимущественно в саркомерных генах) значимо чаще выявлялись у больных с НКМ в сравнении с прочими больными с ДКМП. Наличие НКМ сопровождается более агрессивными желудочковыми нарушениями ритма: значимо более частой желудочковой экстрасистолией, большей частотой оправданных срабатываний дефибрилляторов и значимо более высокой частотой шоков/внезапных сердечных смертей. Частота тромбозов у больных с НКМ была незначимо выше, частота тромбоэмболических осложнений не отличалась от остальных больных с ДКМП благодаря значимо более частому назначению непрямых антикоагулянтов (при меньшей частоте МА). Частота смертей и трансплантаций у больных с НКМ значимо не отличалась от остальных пациентов с ДКМП – вероятно, за счет своевременной профилактики внезапной смерти, эмболических осложнений и положительного ответа на комплексное лечение с достижением удовлетворительного уровня ФВ. Ни в одном случае не отмечено регресса НКМ на фоне улучшения сократимости и сокращения размеров левого желудочка.
Отношения и Деятельность. Нет.
Relationships and Activities. None.
Финансирование. Исследование проведено при поддержке Сеченовского Университета.
Funding. The study was performed with the support of the Sechenov University.
1. Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29(2):270-6. DOI:10.1093/eurheartj/ehm342.
2. Поляк М.Е., Мершина Е.А., Заклязьминская Е.В. Некомпактный миокард левого желудочка: симптом, синдром или вариант развития? Российский Кардиологический Журнал. 2017;(2):106-13. DOI:10.15829/1560-4071-2017-2-106-113.
3. Arbustini E, Favalli V, Narula N, et al. Left Ventricular Noncompaction: A Distinct Genetic Cardiomyopathy? J Am Coll Cardiol. 2016;68(9):949-66. DOI:10.1016/j.jacc.2016.05.096.
4. Hershberger RE, Morales A, Cowan J. Is Left Ventricular Noncompaction a Trait, Phenotype, or Disease? The Evidence Points to Phenotype. Circ Cardiovasc Genet. 2017;10(6):e001968. DOI:10.1161/CIRCGENETICS.117.001968.
5. Pinto YM, Elliott PM, Arbustini E, et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J. 2016;37(23):1850-8. DOI:10.1093/eurheartj/ehv727.
6. Павленко Е.В., Благова О.В., Вариончик Н.В. и др. Регистр взрослых больных с некомпактным миокардом левого желудочка: классификация клинических форм и проспективная оценка их прогрессирования. Российский Кардиологический Журнал. 2019;(2):12-25. DOI:10.15829/1560-4071-2019-2-12-25.
7. Richards S, Aziz N, Bale S, et al.; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-24. DOI:10.1038/gim.2015.30.
8. Jefferies JL, Wilkinson JD, Sleeper LA, et al. Pediatric Cardiomyopathy Registry Investigators. Cardiomyopathy Phenotypes and Outcomes for Children With Left Ventricular Myocardial Noncompaction: Results From the Pediatric Cardiomyopathy Registry. J Card Fail. 2015;21(11):877-84. DOI:10.1016/j.cardfail.2015.06.381.
9. Biagini E, Ragni L, Ferlito M, et al. Different types of cardiomyopathy associated with isolated ventricular noncompaction. Am J Cardiol. 2006;98(6):821-4. DOI:10.1016/j.amjcard.2006.04.021.
10. Towbin JA, Lorts A, Jefferies JL. Left ventricular non-compaction cardiomyopathy. Lancet. 2015;386(9995):813-25. DOI:10.1016/S0140-6736(14)61282-4.
11. van Waning JI, Moesker J, Heijsman D, et al. Systematic Review of Genotype-Phenotype Correlations in Noncompaction Cardiomyopathy. J Am Heart Assoc. 2019;8(23):e012993. DOI:10.1161/JAHA.119.012993.
12. Peters F, Khandheria BK, Botha F, et al. Clinical outcomes in patients with isolated left ventricular noncompaction and heart failure. J Card Fail. 2014;20(10):709-15. DOI:10.1016/j.cardfail.2014.07.007.
13. Oechslin E, Jenni R. Left Ventricular Noncompaction: From Physiologic Remodeling to Noncompaction Cardiomyopathy. J Am Coll Cardiol. 2018;71(7):723-6. DOI:10.1016/j.jacc.2017.12.031.
14. Probst S, Oechslin E, Schuler P, et al. Sarcomere gene mutations in isolated left ventricular noncompaction cardiomyopathy do not predict clinical phenotype. Circ Cardiovasc Genet. 2011;4(4):367-74. DOI:10.1161/CIRCGENETICS.110.959270.
15. Kayvanpour E, Sedaghat-Hamedani F, Gi WT, et al. Clinical and genetic insights into non-compaction: a meta-analysis and systematic review on 7598 individuals. Clin Res Cardiol. 2019;108(11):1297-308. DOI:10.1007/s00392-019-01465-3.
16. Stöllberger C, Blazek G, Dobias C, et al. Frequency of stroke and embolism in left ventricular hypertrabeculation/noncompaction. Am J Cardiol. 2011;108(7):1021-3. DOI:10.1016/j.amjcard.2011.05.039.
17. Kido K, Guglin M. Anticoagulation Therapy in Specific Cardiomyopathies: Isolated Left Ventricular Noncompaction and Peripartum Cardiomyopathy. J Cardiovasc Pharmacol Ther. 2019;24(1):31-6. DOI:10.1177/1074248418783745.
18. Amzulescu MS, Rousseau MF, Ahn SA, et al. Prognostic Impact of Hypertrabeculation and Noncompaction Phenotype in Dilated Cardiomyopathy: A CMR Study. JACC Cardiovasc Imaging. 2015;8(8):934-46. DOI:10.1016/j.jcmg.2015.04.015.
19. Sedaghat-Hamedani F, Haas J, Zhu F, et al. Clinical genetics and outcome of left ventricular noncompaction cardiomyopathy. Eur Heart J. 2017;38(46):3449-60. DOI:10.1093/eurheartj/ehx545.
Благова Ольга Владимировна.
Москва.
Павленко Екатерина Вадимовна.
Москва.
Вариончик Надежда Васильевна.
Москва.
Седов Всеволод Парисович.
Москва.
Гагарина Нина Владимировна.
Москва.
Мершина Елена Александровна.
Москва.
Поляк Маргарита Евгеньевна.
Москва.
Заклязьминская Елена Валерьевна.
Москва.
Недоступ Александр Викторович.
Москва.
Благова О.В., Павленко Е.В., Вариончик Н.В., Седов В.П., Гагарина Н.В., Мершина Е.А., Поляк М.Е., Заклязьминская Е.В., Недоступ А.В. Некомпактный миокард с дилатационным фенотипом: проявления, лечение и исходы в сравнении другими формами синдрома дилатационной кардиомиопатии. Рациональная Фармакотерапия в Кардиологии. 2022;18(1):27-35. https://doi.org/10.20996/1819-6446-2022-02-01
Blagova O.V., Pavlenko E.V., Varionchik N.V., Sedov V.P., Gagarina N.V., Mershina E.A., Polyak M.E., Zaklyazminskaya E.V., Nedostup A.V. Noncompact Myocardium with Dilated Phenotype: Manifestations, Treatment and Outcomes in Comparison with Other Forms of Dilated Cardiomyopathy Syndrome. Rational Pharmacotherapy in Cardiology. 2022;18(1):27-35. https://doi.org/10.20996/1819-6446-2022-02-01


Главный редактор
 Драпкина О. М.
Драпкина О. М.
101990, Москва, Петроверигский пер., 10,
Тел.: +7 (499) 553 68 10
е-mail: otsec.rfc@mail.ru








































