



Содержание
Перейти к:
https://doi.org/10.20996/1819-6446-2024-3005
EDN: OVXPMH
Перейти к:
В статье представлено описание клинического наблюдения пациента молодого возраста с ранее недиагностированной бивентрикулярной аритмогенной кардиомиопатией (АКМП), госпитализированного с подозрением на острый коронарный синдром на фоне эпизода желудочковой тахикардии (ЖТ). На основании клинической картины, повышения тропонина I и наличия зон нарушения локальной сократимости левого желудочка с нарушением его систолической функции при отсутствии значимого атеросклеротического поражения коронарных артерий по данным коронарографии, первоначально был диагностирован инфаркт миокарда (ИМ) 2 типа. Для уточнения генеза ЖТ и характера поражения желудочков пациенту была проведена магнитно-резонансная томография (МРТ). Выявленные при МРТ аномалии правого и левого желудочков, а также результаты генетического исследования позволили верифицировать бивентрикулярную АКМП. Постановка данного диагноза заставила пересмотреть верифицированный ранее ИМ 2 типа в пользу острого повреждения миокарда в рамках патогенеза АКМП. АКМП следует включать в спектр заболеваний для дифференциального диагноза при подозрении на острый миокардит или ИМ у пациентов с повышением тропонина и без значимой обструкции коронарных артерий.
Ставцева Ю.В., Мершина Е.А., Лобжанидзе Т.В., Баздырева Е.И., Хуцишвили Н.И., Кобалава Ж.Д. Бивентрикулярная аритмогенная кардиомиопатия под маской инфаркта миокарда у пациента молодого возраста. Рациональная Фармакотерапия в Кардиологии. 2024;20(1):57-62. https://doi.org/10.20996/1819-6446-2024-3005. EDN: OVXPMH
Stavtseva Y.V., Mershina E.A., Lobzhanidze T.V., Bazdireva E.I., Khutsishvili N.I., Kobalava Zh.D. Acute myocardial infarction-like event in a young patient with biventricular arrhythmogenic cardiomyopathy. Rational Pharmacotherapy in Cardiology. 2024;20(1):57-62. (In Russ.) https://doi.org/10.20996/1819-6446-2024-3005. EDN: OVXPMH
Генетически детерминированная аритмогенная кардиомиопатия (АКМП) является редким заболеванием, характеризующимся фиброзно-жировым замещением миокарда правого (правожелудочковая АКМП), левого (левожелудочковая АКМП) или обоих желудочков (бивентрикулярная АКМП) [1-4]. Прогрессирующее течение заболевания ассоциировано с высоким риском внезапной сердечной смерти (ВСС), желудочковых аритмий и сердечной недостаточности (СН) [4]. Диагностика АКМП представляет затруднения в связи с отсутствием патогномоничных симптомов и признаков. Для улучшения верификации данного заболевания (включая его левожелудочковые фенотипы) в 2020 г. европейские эксперты приняли консенсусный документ с обновленными критериями АКМП (на основе модифицированных ITF критериев 2010 г.), получившими название новые "критерии Падуи". На основании совокупности представленных в документе больших и малых критериев устанавливается диагноз определенной, вероятной или возможной АКМП [2][3]. При постановке диагноза важно также учитывать, что прогрессирование заболевания может характеризоваться эпизодами острого миокардиального повреждения, клинически мимикрирующими под острый инфаркт миокарда (ИМ) или миокардит [4-7]. Поэтому АКМП следует включать в спектр заболеваний для дифференциального диагноза при подозрении на острый миокардит или ИМ у пациентов без значимой обструкции коронарных артерий.
В данной публикации представлено описание клинического наблюдения молодого пациента, госпитализированного с подозрением на острый коронарный синдром на фоне эпизода желудочковой тахикардии (ЖТ), у которого впоследствии была диагностирована АКМП.
Пациент 1991 г.р. (32 года), без вредных привычек, с неотягощенным семейным анамнезом, контролируемой артериальной гипертонией и ожирением 2 степени. По данным анамнеза, в 2010 г. (19 лет) при прохождении медицинской комиссии в военкомате по данным холтеровского мониторирования электрокардиограммы были выявлены частая желудочковая полиморфная экстрасистолия (ЖЭС; 3-6 тыс./сут), неустойчивые пробежки ЖТ (морфология неизвестна). При трансторакальной эхокардиографии (ЭхоКГ) обращали на себя внимание дилатация правых отделов сердца, пролапс митрального клапана (ПМК) до 5 мм. На основании выявленных изменений был диагностирован миокардитический кардиосклероз, ПМК. От дальнейшего обследования (магнитно-резонансная томография (МРТ) сердца, эндоваскулярное электрофизиологическое исследование) пациент отказался. В октябре 2022 г. (31 год) на фоне психоэмоциональных перегрузок пациент почувствовал резкую общую слабость, головокружение, учащенное сердцебиение, дискомфорт за грудиной. Бригадой скорой медицинской помощи на ЭКГ была зарегистрирована мономорфная ЖТ, предположительно левожелудочковой локализации на основании морфологического анализа комплекса QRS (рис. 1) [8, 9]. На фоне внутривенной инфузии амиодарона был восстановлен синусовый ритм. Пациент был госпитализирован в стационар. На ЭКГ при поступлении регистрировались синусовый ритм, атриовентрикулярная блокада 1 степени, признаки рубцовых изменения боковой стенки левого желудочка (ЛЖ), неспецифическая задержка внутрижелудочкового проведения, сглаженный Т зубец в большинстве отведений, эпсилон-волна в грудных отведениях (рис. 2). При ЭхоКГ отмечалось снижение фракции выброса (ФВ) ЛЖ до 36% (акинез базальных отделов нижнего и нижне-перегородочного сегментов, гипокинез среднего и апикального сегментов боковой стенки с переходом на верхушку с формированием аневризмы) без его дилатации [индексированный конечно-диастолический объем (КДО) ЛЖ 85 мл/м2, конечно-систолический объем ЛЖ 42 мл/м2]. Признаков ПМК нет. Выявлена дилатация правого желудочка (ПЖ; передне-задний размер 39 мм, базальный — 48 мм, средний — 38 мм, диаметр выходного тракта ПЖ по длинной оси 45 мм) с нарушением его сократимости и систолической функции (диффузный гипокинез, фракция изменения площади 20%, TAPSE 12 мм); систолическое давление в легочной артерии 30 мм рт.ст., скорость трикуспидальной регургитации 2,7 мс) (рис. 3). В крови определялся повышенный уровень тропонина I до 7,5 нг/мл с последующим снижением до нормальных значений. Других изменений в лабораторных исследованиях (электролиты, гормоны щитовидной железы, воспалительные маркеры, вирусные и иммунные тесты) выявлено не было. При коронароангиографии гемодинамически значимых поражений коронарных артерий не обнаружено. При холтеровском мониторировании ЭКГ за весь период отмечался синусовый ритм, 400 (0,39% в сут) мономорфных ЖЭС. С учетом клинической картины, динамики тропонина I, зон нарушения локальной сократимости ЛЖ и отсутствия изменений при коронароангиографии первоначально был установлен диагноз инфаркта миокарда 2 типа. Назначена двойная антиагрегантная терапия. Для уточнения характера поражения желудочков и генеза желудочковой аритмии пациенту была проведена МРТ: выявлено рубцовое поражение миокарда ЛЖ в базальных боковых сегментах с признаками отека, умеренное снижение сократительной функции ЛЖ (49%) без дилатации его полости; признаки выраженной дилатации полости ПЖ (индексированный КДО ПЖ 152 мл/м2), снижение сократимости (ФВ ПЖ 25%). Обнаружены участки интрамиокардиального фиброза миокарда правого и левого желудочков некоронарогенной природы (рис. 4).
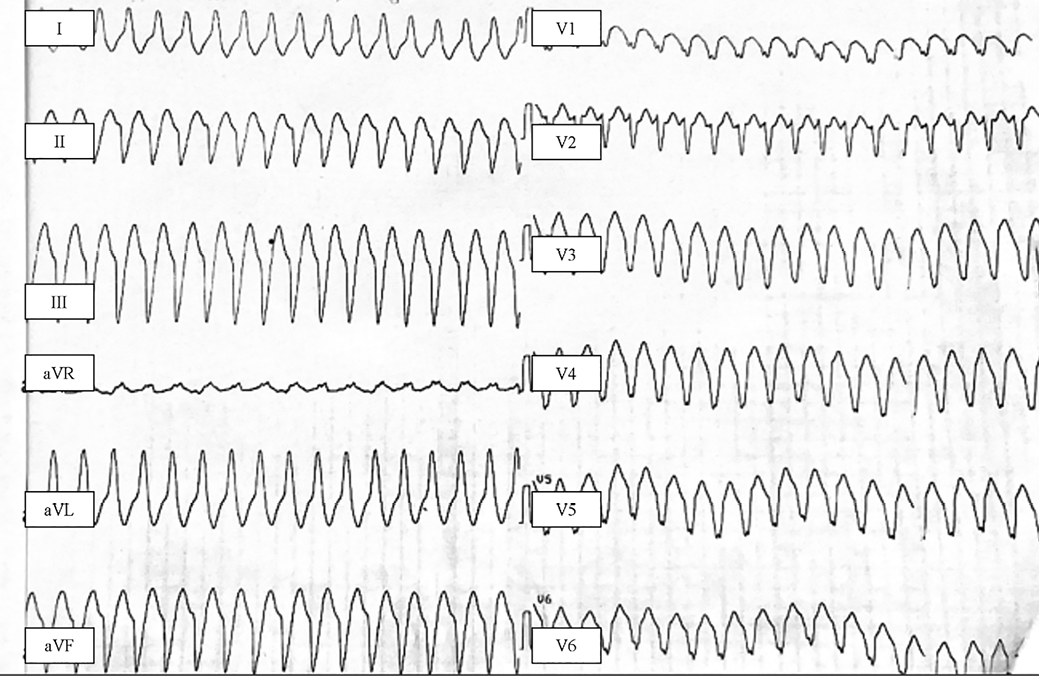
Рис. 1. Электрокардиограмма во время эпизода ЖТ (25 мм/с, 10 мм/мВ): мономорфная тахикардия с широкими комплексами и частотой 188 уд/мин.
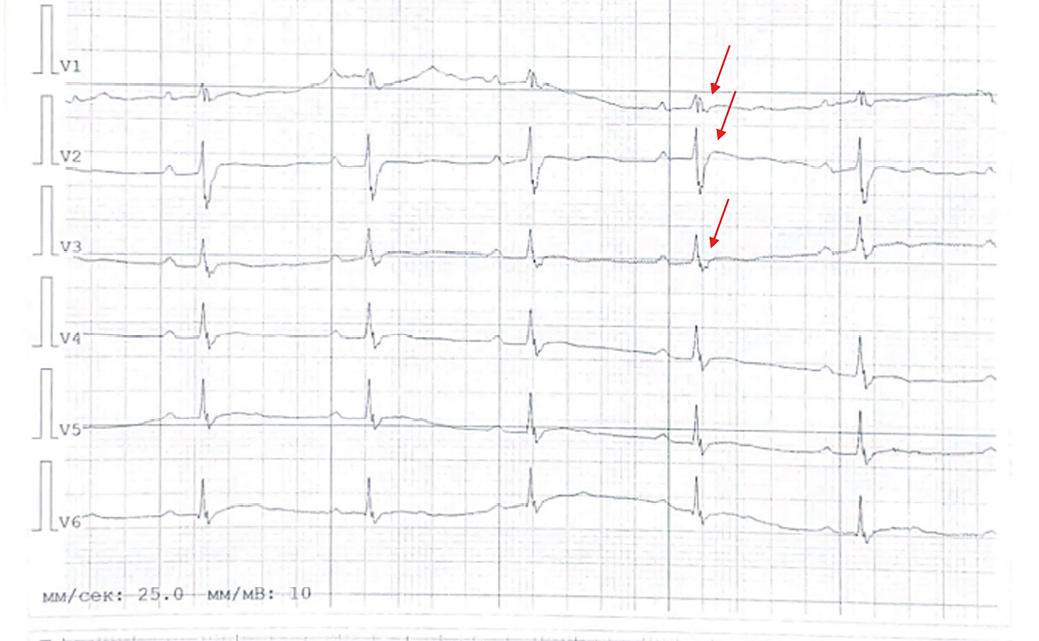
Рис. 2. Электрокардиограмма (25 мм/с, 10 мм/мВ): синусовый ритм с ЧСС 60 уд./мин, отклонение электрической оси сердца вправо. Атриовентрикулярная блокада 1 степени, рубцовые изменения боковой стенки левого желудочка, неспецифическая задержка внутрижелудочкового проведения. Эпсилон-волна (красные стрелки). Сглаженный Т зубец в большинстве отведений.
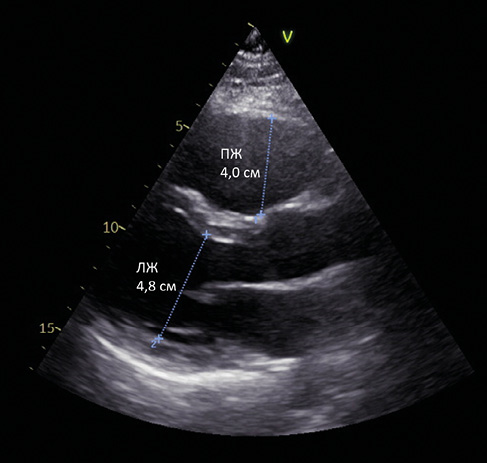
Рис. 3. Эхокардиограмма. Парастернальный доступ, длинная ось левого желудочка. Увеличение передне-заднего размера правого желудочка. Левый желудочек не увеличен.
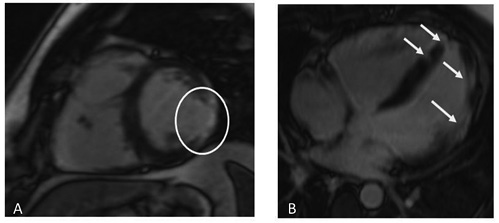
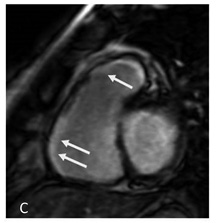
Рис. 4. МРТ — изображения с отсроченным контрастированием демонстрируют субэндокардиальный участок накопления контраста в среднем сегменте заднебоковой стенки ЛЖ по короткой оси (А, обведен белым цветом), отсроченное субэпикардиальное накопление контраста в боковой стенке ЛЖ и межжелудочковой перегородке по длинной оси четырехкамерной проекции (В; белые стрелки); трансмуральное накопление контраста в свободной стенке и в области выходного тракта ПЖ по короткой оси (С, белые стрелки).
Таким образом, при ЭхоКГ и МРТ были выявлены морфофункциональные и структурные изменения правого и левого желудочков сердца, соответствующие большим "критериям Падуи", на основании которых была диагностирована бивентрикулярная форма АКМП [2]. В этой связи ранее описанные клиническая картина, изменения, зарегистрированные по данным лабораторных и инструментальных исследований при поступлении, были интерпретированы в рамках эпизода острого миокардиального повреждения, характерного для данной патологии. Рекомендован прием следующих препаратов: валсартан/сакубитрил 100 мг 2 р/сут, дапаглифлозин 10 мг/сут, эплеренон 25 мг/сут, соталол 80 мг 3 р/сут. Двойная антиагрегантная терапия отменена. Также с целью вторичной профилактики ВСС пациенту рекомендована имплантация кардиовертер-дефибриллятора (ИКД).
В феврале 2023 г. (32 года) зарегистрирован повторный эпизод ЖТ с прежней морфологией без связи с физической и эмоциональной нагрузкой, купированный инфузией амиодарона. При ЭхоКГ клинически значимых динамических изменений не отмечалось. В том же году был имплантирован двухкамерный ИКД. При наблюдении до момента описания данного клинического случая срабатываний устройства не было.
Исследование генов, ассоциированных с АКМП, выявило у пробанда две гетерозиготные мутации на 18 хромосоме гена DSG2: c. 1A>C (p.Met1Leu — start loss в 1 экзоне) и c. 797A>G (p.Asn266Ser — несинонимичная замена в 7 экзоне), определенные предсказательными программами как вероятно патогенная и патогенная соответственно. Кроме того, обнаружена гетерозиготная вероятно патогенная мутация на 4 хромосоме гена ANK2: c.72+2T>C (мутация, нарушающая сайт сплайсинга). Для стратификации риска всем членам семьи пациента был рекомендован каскадный генетический скрининг. На момент описания данного клинического случая его результаты неизвестны.
АКМП представляет собой первичное заболевание миокарда, характеризующееся прогрессирующей гибелью и фиброзно-жировым замещением кардиомиоцитов желудочков вследствие дефектов генов, кодирующих белки десмосом, компоненты цитоскелета и ионных каналов, участвующих в поддержании целостности клеток миокарда и их взаимодействия. В результате формируется субстрат для развития жизнеугрожающих желудочковых аритмий, развития и прогрессирования СН [4, 10, 11].
Следует отметить, что патогенез разрушения кардиомиоцитов на фоне АКМП остается не до конца изученным. Описаны фенотипы АКМП, для которых характерны эпизоды острого повреждения миокарда с массовой гибелью кардиомиоцитов ("горячие фазы") и появлением симптомов миокардита или острого коронарного синдрома, затрудняющих верификацию основного заболевания [5][6][12]. В нашем случае, вероятно, подобные изменения способствовали индукции ЖТ (с предполагаемой локализацией аритмогенного субстрата в ЛЖ) и возникновению клинической картины острого коронарного синдрома, которые первоначально были трактованы в пользу ИМ 2 типа. Однако ранний дебют желудочковой эктопии, характерные морфофункциональные и структурные изменения обоих желудочков (большие критерии АКМП), выявленные при ЭхоКГ и МРТ, отсутствие данных за другую патологию сердца (саркоидоз, амилоидоз, другие кардиомиопатии) убедительно свидетельствовали в пользу бивентрикулярного фенотипа АКМП.
Согласно литературным данным, случаи острого миокардиального повреждения, клинически мимикрирующие под миокардит или острый коронарный синдром, нередко наблюдаются у пациентов с мутациями в генах DSP и DSG2 (чаще леводоминантные и бивентрикулярные типы АКМП) [6][12][13-15]. В нашем случае генетическое исследование выявило гетерозиготные мутации в генах DSG2 и ANK (дигенная мутация). Дигенные мутации обнаруживаются в 4-16% случаев и обусловливают более злокачественный фенотип заболевания, ассоциированный с высоким риском желудочковых аритмий и ВСС в молодом возрасте [16][17].
В соответствии с рекомендациями HRS генетическое исследование является обязательным только для пациентов с изолированной леводоминантной формой АКМП [18]. Однако отрицательный результат генотипирования не исключает того, что АКМП может быть вызвана неизвестной мутацией. В связи с чем экспертная группа HRS рекомендует генотипирование, в первую очередь, как дополнительный этап, повышающий специфичность диагностики АКМП, а также в качестве инструмента стратификации риска и скрининга членов семьи пробанда [18].
Накопленные данные свидетельствуют также о том, что вирусные инфекции или иммунные реакции могут способствовать генетически запрограммированному острому некрозу миокарда [19]. В нашем случае клинических признаков вирусного воспаления или ранее перенесенных инфекций не отмечалось.
Основные терапевтические стратегии при АКМП включают ограничение высокоинтенсивной физической нагрузки, бета-блокаторы, препараты для лечения сердечной недостаточности и профилактики желудочковых нарушений ритма, имплантацию ИКД, катетерную аблацию, в редких случаях — трансплантацию сердца [1][18][20]. В нашем случае после имплантации ИКД на фоне медикаментозной терапии срабатываний устройства при длительности наблюдения в течение 9 месяцев зафиксировано не было. Однако АКМП является прогрессирующим заболеванием, при котором на фоне фиброзного замещения миокарда желудочков могут возникать условия для рецидивирования ЖТ. При данном сценарии следует рассмотреть вопрос о проведении эндоваскулярного электрофизиологического исследования и катетерной аблации аритмогенного субстрата для предотвращения срабатываний ИКД и улучшения качества жизни больного.
Некоторые генетические варианты АКМП могут иметь фенотипы сходные с миокардитом и острым коронарным синдромом. В связи с этим, в отсутствие патогномоничных признаков и симптомов, а также идеального метода диагностики, АКМП следует рассматривать при проведении дифференциального диагноза с представленными выше состояниями, особенно у пациентов с увеличением уровня тропонина, интактными коронарными артериями и бивентрикулярной кардиомиопатией неясной этиологии.
Отношения и Деятельность. Нет.
Relationships and Activities. None.
1. Bennett RG, Haqqani HM, Berruezo A, et al. Arrhythmogenic Cardiomyopathy in 2018-2019: ARVC/ALVC or Both? Heart Lung Circ. 2019;28(1):164-177. DOI:10.1016/j.hlc.2018.10.013.
2. Corrado D, Perazzolo Marra M, Zorzi A, et al. Diagnosis of arrhythmogenic cardiomyopathy: The Padua criteria. Int J Cardiol. 2020;319(15):106-114. DOI:10.1016/j.ijcard.2020.06.005.
3. Miles C, Finocchiaro G, Papadakis M, et al. Sudden Death and Left Ventricular Involvement in Arrhythmogenic Cardiomyopathy. Circulation. 2019;139(15):1786-1797. DOI:10.1161/CIRCULATIONAHA.118.037230.
4. Sen-Chowdhry S, Syrris P, Prasad SK, et al. Left-dominant arrhythmogenic cardiomyopathy: an under-recognized clinical entity. J Am Coll Cardiol. 2008;52(25):2175-87. DOI:10.1016/j.jacc.2008.09.019.
5. Patrianakos AP, Protonotarios N, Nyktari E, et al. Arrhythmogenic right ventricular cardiomyopathy/dysplasia and troponin release. Myocarditis or the “hot phase” of the disease? Int J Cardiol. 2012;157(2):e26-8. DOI:10.1016/j.ijcard.2011.09.017.
6. Bariani R, Cipriani A, Rizzo S, et al. ‘Hot phase’ clinical presentation in arrhythmogenic cardiomyopathy. Europace. 2021;23(6):907-917. DOI:10.1093/europace/euaa343.
7. Smith ED, Lakdawala NK, Papoutsidakis N, et al. Desmoplakin Cardiomyopathy, a Fibrotic and Inflammatory Form of Cardiomyopathy Distinct From Typical Dilated or Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation. 2020;141(23):1872-1884. DOI:10.1161/CIRCULATIONAHA.119.044934.
8. Kashou AH, Noseworthy PA, DeSimone CV. Wide Complex Tachycardia Differentiation: A Reappraisal of the State-of-the-Art. J Am Heart Assoc. 2020;9(11):e016598. DOI:10.1161/JAHA.120.016598.
9. Barold SS, Stroobandt RX, Herweg B. Limitations of the negative concordance pattern in the diagnosis of broad QRS tachycardia. J Electrocardiol. 2012;45(6):733-5. DOI:10.1016/j.jelectrocard.2012.06.017.
10. Corrado D, Basso C, Judge DP. Arrhythmogenic Cardiomyopathy. Circ Res. 2017;121(7):784-802. DOI:10.1161/CIRCRESAHA.117.309345.
11. Austin KM, Trembley MA, Chandler SF, et al. Molecular mechanisms of arrhythmogenic cardiomyopathy. Nat Rev Cardiol. 2019;16(9):519-537. DOI:10.1038/s41569-019-0200-7.
12. Lota AS, Hazebroek MR, Theotokis P, et al. Genetic Architecture of Acute Myocarditis and the Overlap With Inherited Cardiomyopathy. Circulation. 2022;146(15):1123-1134. DOI:10.1161/CIRCULATIONAHA.121.058457.
13. Lopez-Ayala JM, Pastor-Quirante F, Gonzalez-Carrillo J, et al. Genetics of myocarditis in arrhythmogenic right ventricular dysplasia. Heart Rhythm. 2015;12(4):766-73. DOI:10.1016/j.hrthm.2015.01.001.
14. Brodehl A, Meshkov A, Myasnikov R, et al. Hemi- and Homozygous Loss-ofFunction Mutations in DSG2 (Desmoglein-2) Cause Recessive Arrhythmogenic Cardiomyopathy with an Early Onset. Int J Mol Sci. 2021;22(7):3786. DOI:10.3390/ijms22073786.
15. Brodehl A, Weiss J, Debus JD, et al. A homozygous DSC2 deletion associated with arrhythmogenic cardiomyopathy is caused by uniparental isodisomy. J Mol Cell Cardiol. 2020;141:17-29. DOI:10.1016/j.yjmcc.2020.03.006.
16. Bhonsale A, Groeneweg JA, James CA, et al. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathyassociated mutation carriers. Eur Heart J. 2015;36(14):847-55. DOI:10.1093/eurheartj/ehu509.
17. Rigato I, Bauce B, Rampazzo A, et al. Compound and digenic heterozygosity predicts lifetime arrhythmic outcome and sudden cardiac death in desmosomal gene-related arrhythmogenic right ventricular cardiomyopathy. Circ Cardiovasc Genet. 2013;6(6):533-42. DOI:10.1161/CIRCGENETICS.113.000288.
18. Towbin JA, McKenna WJ, Abrams DJ, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm. 2019;16(11):e301-e372. DOI:10.1016/j.hrthm.2019.05.007.
19. Asatryan B, Asimaki A, Landstrom AP, et al. Inflammation and Immune Response in Arrhythmogenic Cardiomyopathy: State-of-the-Art Review. Circulation. 2021;144(20):1646-1655. DOI:10.1161/CIRCULATIONAHA.121.055890.
20. Lie ØH, Dejgaard LA, Saberniak J, et al. Harmful Effects of Exercise Intensity and Exercise Duration in Patients With Arrhythmogenic Cardiomyopathy. JACC Clin Electrophysiol. 2018;4(6):744-753. DOI:10.1016/j.jacep.2018.01.010.
Ставцева Юлия Вадимовна
Москва
Мершина Елена Александровна
Москва
Лобжанидзе Тинатин Викторовна
Москва
Баздырева Елена Ивановна
Москва
Хуцишвили Нуцико Ивановна
Москва
Кобалава Жанна Давидовна
Москва
Ставцева Ю.В., Мершина Е.А., Лобжанидзе Т.В., Баздырева Е.И., Хуцишвили Н.И., Кобалава Ж.Д. Бивентрикулярная аритмогенная кардиомиопатия под маской инфаркта миокарда у пациента молодого возраста. Рациональная Фармакотерапия в Кардиологии. 2024;20(1):57-62. https://doi.org/10.20996/1819-6446-2024-3005. EDN: OVXPMH
Stavtseva Y.V., Mershina E.A., Lobzhanidze T.V., Bazdireva E.I., Khutsishvili N.I., Kobalava Zh.D. Acute myocardial infarction-like event in a young patient with biventricular arrhythmogenic cardiomyopathy. Rational Pharmacotherapy in Cardiology. 2024;20(1):57-62. (In Russ.) https://doi.org/10.20996/1819-6446-2024-3005. EDN: OVXPMH
.jpg)
Главный редактор
 Драпкина О. М.
Драпкина О. М.
101990, Москва, Петроверигский пер., 10,
Тел.: +7 (499) 553 68 10
е-mail: otsec.rfc@mail.ru







































