



Содержание
Перейти к:
https://doi.org/10.20996/1819-6446-2024-3022
EDN: JNMVKN
Перейти к:
Появление ингибиторов иммунных контрольных точек (ИКТ) открыло новые перспективы в иммунотерапии рака. Тем не менее, серьезные, в том числе опасные для жизни состояния, вызванные кардиотоксическими эффектами ИКТ, ставят перед клиническими специалистами ряд препятствий. Нехватка знаний об аспектах патофизиологии кардиоваскулярных нежелательных явлений терапии опухолей ИКТ является одной из причин обращения специалистов онкологического профиля за помощью к кардиологам. В РФ данная проблема является еще более актуальной в связи с тем, что в научной литературе не имеется первичных исследований, которые были бы посвящены детальному разбору вопросов патогенеза кардиоваскулярных нарушений, возникающих в ходе терапии ингибиторами ИКТ. В зарубежной литературе представлены работы, которые рассматривают механизмы развития отдельных осложнений, но число работ, которые бы систематизировали и суммировали описания всех наиболее значимых осложнений терапии ингибиторами ИКТ, невелико. В связи с этим был проведен обзор литературы по применению ингибиторов ИКТ с поиском в библиографических базах данных PubMed, Embase, Web of Scienсe, e- Library, Google Scholar, целью которого стал анализ накопленных данных о механизмах развития осложнений терапии ингибиторами ИКТ; предпочтение отдавалось систематическим обзорам, рандомизированным клиническим исследованиям, которые были бы дополнены отдельными когортными исследованиями и описанием некоторых экспериментов. Таким образом, определено, что кардиотоксичность ингибиторов ИКТ может затрагивать любой отдел сердечно-с осудистой системы, вызывая изменения как воспалительной (миокардит, перикардит, коронарный васкулит), так и невоспалительной этиологии (аритмии, невоспалительная дисфункция левого желудочка, такоцубоподобный синдром, коронарный вазоспазм и инфаркт миокарда на фоне атеросклероза). За счет разнообразия клинических проявлений, при относительной редкости, кардиотоксические реакции ингибиторов ИКТ могут создавать трудности при дифференциальной диагностике и лечении пациентов. Понимание же их механизмов повышает возможности специалистов в отношении формирования эффективной стратегии лечения с минимизацией риска осложнений. И хотя в онкологической практике накоплено немало теоретических, экспериментальных и клинико-э мпирических данных о побочных эффектах этого класса противоопухолевых препаратов, кардиотоксичность ингибиторов ИКТ представляет собой проблему, требующую дальнейших исследований.
Хидирова Л.Д., Лацвиева А.Е., Ведерин А.А. Механизмы кардиотоксичности противоопухолевой терапии ингибиторами иммунных контрольных точек: современные достижения. Рациональная Фармакотерапия в Кардиологии. 2024;20(2):265-274. https://doi.org/10.20996/1819-6446-2024-3022. EDN: JNMVKN
Khidirova L.D., Latsvieva A.E., Vederin A.V. Cardiotoxicity mechanisms of antitumor therapy with immune checkpoint inhibitors: new achievements. Rational Pharmacotherapy in Cardiology. 2024;20(2):265-274. (In Russ.) https://doi.org/10.20996/1819-6446-2024-3022. EDN: JNMVKN
Революционным событием в иммунотерапии онкологических заболеваний стало создание ингибиторов иммунных контрольных точек (ИКТ), которые представляют гуманизированные моноклональные антитела к ингибирующим рецепторам и их лигандам, повышающим эффективность Т-клеточного иммунного ответа. Однако чрезмерная его активация приводит к уникальным побочным эффектам — иммуноопосредованным нежелательным реакциям (иоНР) [1][2]. В их числе находится и кардиотоксичность, которая встречается достаточно редко, у 1,1-5,0% пациентов, однако смертность достигает от 27 до 50% случаев [3]. Очевидно, что кардиотоксичность ингибиторов ИКТ требует особой клинической настороженности. Понимание основных клинико-диагностических предпосылок кардиотоксических осложнений ингибиторов ИКТ и разработка протоколов ведения таких пациентов, несомненно, должны иметь в своей основе информацию о патогенетических механизмах этих реакций. В этой связи цель данной работы — описание по данным литературы некоторых базовых аспектов кардиотоксических эффектов ингибиторов ИКТ.
На сегодняшний день одобрено три класса ингибиторов ИКТ, отличающихся по воздействию на ИКТ [4]:
CTLA-4 — белок, экспрессирующийся на поверхности регуляторных (Тreg) и других Т-лимфоцитов после их активации. Он гомологичен CD28, стимулирующему ко-рецептору, необходимому для активации покоящихся Т-лимфоцитов при взаимодействии поверхностного белкового комплекса Т-лимфоцитов — Т-клеточного рецептора (TCR) и специфических пептидов опухоли, представляемых в составе молекулы главного класса гистосовместимости (MHC) антиген-презентирующими клетками (АПК) в лимфоузлах. В отличие от CD28, CTLA-4 передает ко-ингибирующий сигнал, один из механизмов его передачи — конкуренция CTLA-4 с CD28 за связывание с CD80/86 на АПК. Передача ингибирующего сигнала приводит к анергии Т-лимфоцитов, как при взаимодействии непосредственно с АПК, так и опосредованно через Treg, рекрутируемых на фоне длительного воспаления. Это приводит к подавлению противоопухолевого иммунитета на уровне лимфоузлов, дренирующих опухолевые очаги [5]. Ингибирование рецепторов CTLA-4 блокирует эти ингибирующие сигналы и усиливает активацию Т-клеток и противоопухолевый ответ (рис. 1).
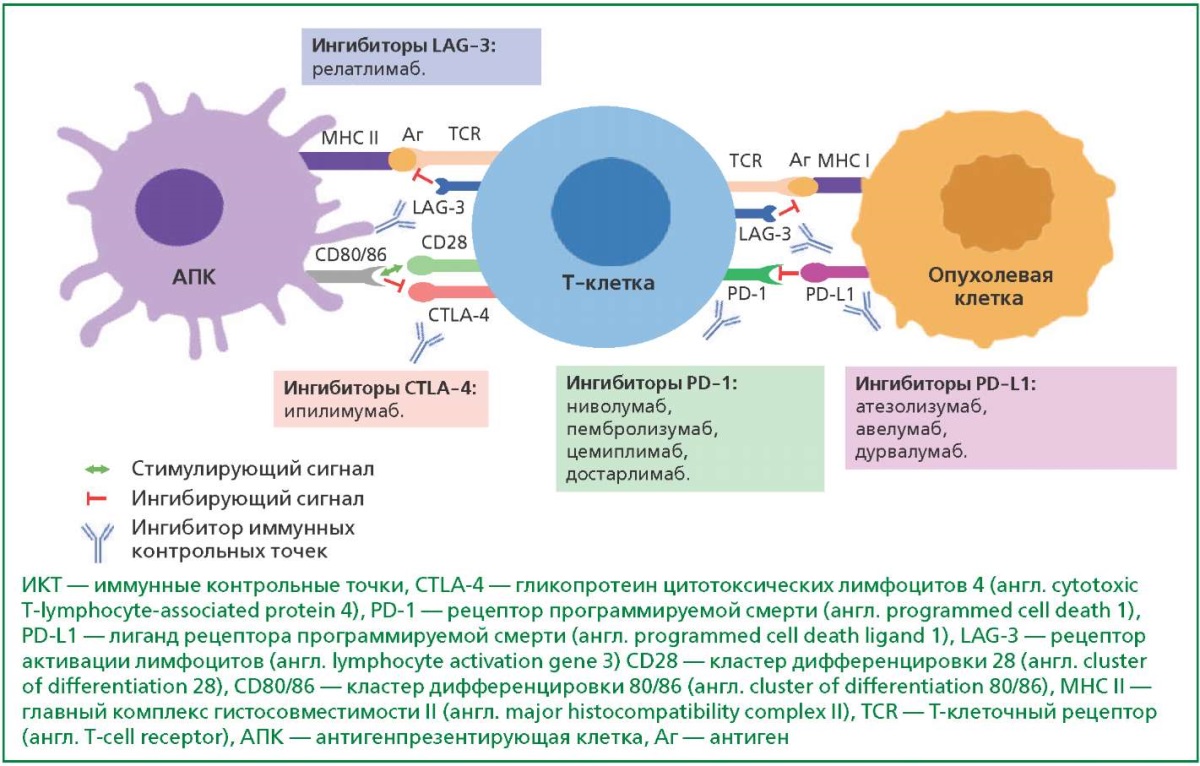
Рисунок 1. Механизм активации Т-клеток и ИКТ как мишени воздействия ингибиторов ИКТ
PD-L1 — это белок клеток-мишеней, связывающийся с рецепторами PD-1 на Т-клетках CD8, ограничивающий воспаление и предотвращающий повреждение здоровых тканей. PD-1 экспрессируется на активированных Т- и В-лимфоцитах, NK-клетках и макрофагах, причем высокая экспрессия PD-1 наблюдается у "истощенных" Т-лимфоцитов, ранее подвергнутых высокому уровню антигенной стимуляции. PD-L1 экспрессируется на гемопоэтических клетках (например, АПК), а также на клетках различных тканей, в том числе на структурах сердца. Ось PD-1/PD-L1 играет важную роль в поддержании толерантности иммунитета к собственным тканям организма: при активации аутореактивных Т-лимфоцитов экспрессия PD-L1 на АПК и клетках ткани приводит к связыванию PD-L1 с PD-1 и ингибированию сигнала, генерируемого TCR, с последующим апоптозом Т-лимфоцитов. Экспрессия PD-L1 на клетках опухоли и АПК, усиливаемая ИФН-γ и другими цитокинами эффекторных Т-лимфоцитов, предотвращает активацию Т-клеток и позволяет опухоли уклоняться от иммунного ответа [5]. Ингибиторы ИКТ, нацеленные на рецепторы PD-1 или PD-L1, предотвращают ускользание опухоли путем сверхэкспрессии PD-L1, что приводит к цитотоксическому разрушению опухолевых клеток, опосредованному CD8 Т-клетками (cм. рис. 1).
LAG-3 — ко-ингибирующий белок, уникально присутствующий как на цитотоксических Т-клетках CD8, так и на иммуносупрессивных регуляторных Т-клетках. LAG-3 связывается с комплексом ТCR:CD3 на мембране Т-клеток и блокирует передачу сигнала TCR, что прекращает пролиферацию клеток и секрецию цитокинов в ответ на передачу сигналов CD3. Совместное взаимодействие LAG-3 и CD3 в иммунологическом синапсе необходимо для понижения модуляции передачи сигнала TCR. Среди других последствий одновременное взаимодействие LAG-3/TCR с их лигандами ингибирует TCR:CD3-зависимые внутриклеточные потоки кальция. Подобно PD-1, конститутивная экспрессия LAG-3 часто связана с истощением Т-клеток [6]. Ингибирование LAG-3 увеличивает ответ Т-клеток CD8 как прямо, так и косвенно за счет снижения регуляторных механизмов Т-клеток (см. рис. 1), с аддитивными противоопухолевыми эффектами в сочетании с ингибированием PD-1.
Активация Т-клеток начинается с презентации опухолевого антигена АПК из врожденной иммунной системы Т-клеткам через МНС, также необходим второй ко-стимулирующий сигнал от связывания CD80 и CD86 на АПК с CD28 на Т-клетках. Внутриклеточный каскад этих сигналов приводит к дифференцировке покоящихся CD8 Т-клеток в активированные цитотоксические CD8 Т-клетки. Нарушение в функционировании CTLA-4, LAG-3 и оси PD-1/PD-L1, возникающее в результате применения ингибиторов ИКТ, приводит к нарушению механизмов, поддерживающих периферическую иммунологическую толерантность к здоровым тканям и органам [5].
Миокардит — наиболее распространенная кардиотоксическая иоНР. Хотя среди всех системных иоНР его встречаемость составляет около 1,14%, летальность при развитии этого осложнения высокая — около 25-50% [7]. В силу клинической актуальности данное осложнение наиболее изучено. Миокардит может сформироваться уже через 2 недели после терапии ингибиторами ИКТ и протекать как бессимптомно с возрастанием уровня сердечных биомаркеров, так и с тяжелыми поражениями сердца, вплоть до молниеносных и опасных для жизни проявлений: кардиогенного шока, сердечной недостаточности, жизнеугрожающих аритмий. Для правильного формирования стратегий диагностики, профилактики и лечения ИКТ-ассоциированного миокардита требуется знание его патогенетических механизмов, имеющих несколько взаимосвязанных аспектов [7].
В фундаментальной основе патогенеза миокардита лежат пути CTLA-4 и PD-1/PD-L1. В норме они играют важную роль в установлении периферической иммунной толерантности к структурам сердца, защищая миокард от повреждений, как аутоиммунного, так и неаутоиммунного характера; например, ось PD-1/PD-L1 выступает механизмом защиты миокарда в случае его ишемического поражения [8]. Блокада путей CTLA-4 и PD-1/PD-L1 в результате терапии таргетными препаратами приводит к Т-клеточной агрессии, что было продемонстрировано в доклинических исследованиях на мышиных моделях. В работе S. C. Wei и соавт. было показано спонтанное развитие миокардита при ингибировании CTLA-4 в эксперименте с применением анти-CTLA-4 препаратов, что связывалось со снижением активности Treg, экспрессирующих CTLA-4, приводящей к усиленной активации кардиореактивных Т-клеток [9]. Также была продемонстрирована роль делеции CTLA-4 в развитии фатальной неконтролируемой лимфопролиферации, диффузного воспаления тканей, включая миокардит, и преждевременной смертности [10].
Экспериментально же вносимые дефекты функционирования оси PD-1/PD-L1 приводили к различным сердечным фенотипам, что гипотетически было связано с особенностями генетического фона. Так у мышей, имеющих аутоиммунный генетический фон или имеющих дефицит других контрольных иммунных точек (LAG-3), была показана статистически значимая связь между потерей экспрессии PD-1 и PD-L1/PD-L2 и развитием летального аутоиммунного миокардита, в отличие от исследований с мышами, имеющих неаутоиммунный генетический фон [9-11]. Это свидетельствует о том, что существуют генетические особенности организма, обеспечивающие избыток механизмов регуляции и компенсирующие потерю PD-1, однако в условиях стимулирования иммунной системы нарушение функционирования оси PD-L1/PD-1 может вносить вклад в развитие миокардита даже в отсутствие аутоиммунного генетического фона [11].
Существуют и другие аспекты патогенеза ИКТ-ассоциированного миокардита. Одна из существующих в настоящее время гипотез предполагает его связь с высоким уровнем цитокиновой активности, вызываемой ингибиторами ИКТ. Цитокины, рекрутирующие иммунные клетки в микроокружение опухоли, являются важными модуляторами иммунного ответа, в том числе они имеют отношение и к ИКТ [12]. Например, ось PD-1/PD-L1 является составной частью цитокин-опосредованной петли отрицательной обратной связи, функционирующей в миокарде: секреция ИФН-γ Th1 и цитотоксическими Т-лимфоцитами приводит к повышению активности PD-L1 на эндотелиальных клетках сердца, который в свою очередь приводит к подавлению эффекторных Т-клеток Выключение PD-1/PD-L1 ведет к усилению продукции ИФН-γ и повышению активности Т-клеток, в том числе, в отношении кардиотоксических эффектов [11][13]. Кроме того, ингибиторы ИКТ вызывают повышение уровня других цитокинов. Так, в исследовании S. Y. Lim и соавт., было показано, что на фоне терапии ингибиторами ИКТ имеется повышенная экспрессия 11 цитокинов (G-CSF, GM-CSF, фракталкин, FGF-2, IFNα2, IL12p70, IL1a, IL1B, IL1RA, IL2 и IL13), которая оказалась тесно связана с тяжелыми иоНР [12]. Эти цитокины, обладая мощной провоспалительной активностью, включают стимуляцию рекрутирования Т-клеток, пролиферации, выживания, дифференцировки и эффекторных функций; многие из этих цитокинов (например, IL1a, IL1b, IL2, IFNα2 и IL12p70) участвуют в воспалении, лежащем в основе аутоиммунных процессов. Эти цитокины способствуют проникновению ингибиторов ИКТ в органы-немишени, включая миокард, опосредуя их прямой кардиотоксический Т-лимфоцитопосредованный эффект [13].
Другая гипотеза рассматривает развитие кардиотоксичности вследствие нарушения центральной иммунной толерантности к структурам сердца, которое особенно часто наблюдается при применении ингибиторов ИКТ по поводу опухолей тимуса. Нарушение нормального функционирования вилочковой железы вследствие опухолевого поражения приводит к нарушению стимуляции предшественников Т-клеток антигенами сердца (например, α-миозина с тяжелой цепью) и отрицательного отбора антиген-специфичных Т-клеток миокарда. Впоследствии они созревают в аутореактивные наивные Т-лимфоциты, которые рециркулируют через лимфатические узлы, дренирующие сердце [14]. Нарушение периферической иммунной толерантности, которое возникает при применении ингибиторов ИКТ, может привести к активации сердечных антиген-специфических наивных Т-клеток и их дифференцировке в клоны эффекторных Т-клеток, что дополняет уже имеющееся нарушение в центральной иммунной толерантности к сердцу и в дальнейшем может привести к повреждению миокарда. Это подтверждается тем, что частота развития аутоиммунного миокардита на фоне применения ингибиторов ИКТ выше у пациентов с эпителиальными опухолями тимуса, чем при других опухолях (5-9,1% против 0,06-1%) [15].
В качестве немаловажного антигена, участвующего в развитии аутоиммунного миокардита на фоне применения ингибиторов ИКТ, описан сердечный миозин. В исследовании T. Won и соавт. было показано, что иммунизация миозином или его укороченным пептидом в эксперименте является эффективным методом индуцирования развития аутоиммунного миокардита у различных линий мышей [16]. Была зафиксирована высокая активность субпопуляции миозин-специфических Т-клеток с фенотипом коэкспрессии PD-1, CD44 и CD69, CD44 и CD69, которые являются маркерами активации и имеются на эффекторных, центральных или периферических Т-клетках памяти, уже подвергавшихся антигенной стимуляции. Вместе с миозином были описаны и другие сердечные антигены, такие как β1-адренорецептор, мускариновый рецептор М2 и Na-K-АТФаза, распознаваемые аутоантителами у пациентов с миокардитом и дилатационной кардиомиопатией, что может свидетельствовать об участии нескольких различных аутоиммунных мишеней в патогенезе ИКТ-ассоциированного миокардита [15][17].
В настоящее время высказывается возможность инфильтрации и повреждения миокарда клоном Т-клеток, нацеленным на гомологичный для опухоли и миокарда антиген. Предположение о наличии молекулярной мимикрии между опухолевыми клетками и сердечной тканью косвенно подтверждается экспансией аналогичных клонов Т-клеток в опухолевой ткани и миокарде, выявленной при генотипировании TCR у пациентов, страдающих ИКТ-ассоциированным миокардитом [16][17]. Установлено, что в опухолях пациентов, у которых развивался ИКТ-ассоциированный миокардит, наблюдался высокий уровень мышечно-специфических антигенов (десмин, тропонин) [16]. ИКТ, усиливая Т-клеточный ответ, способствуют активации в том числе и таких аутоагрессивных Т-лимфоцитов, обусловливая развитие перекрестной реакции [18].
Имеются предположения о влиянии окружающей среды на возникновение ИКТ-ассоциированного миокардита. В исследовании C. Gil-Cruz и соавт. есть данные о том, что пептидные миметики тяжелой цепи α-миозина, полученные из комменсальных видов Bacteroides, вызывали прогрессирование аутоиммунного миокардита вплоть до летального исхода экспериментальной модели на мышах посредством активирования Th17 в кишечнике и CD4+ клеток, экспрессирующих специфичный к тяжелой цепи α-миозина TCR. При этом антибактериальная терапия предотвращала летальный исход у мышей [19].
Любопытным представляется механизм антителозависимой клеточно-опосредованной цитотоксичности. Предполагается, что ИКТ, взаимодействуя с CTLA-4, FGL1, LAG-3, PD-1 и PD-L1, могут приводить к опосредованному комплементом повреждению тканей. Fc-область моноклональных антител IgG1 связывается с рецепторами на NK-клетках, собственно, и опосредующих антителозависимую цитотоксичность [18]. И хотя большинство моноклональных антител ИКТ представляют собой IgG4, не опосредующие комплемент-зависимой токсичности, некоторые препараты, например, авелумаб (ингибитор PD-L1), представляют собой моноклональное антитело IgG1. Такие ингибиторы ИКТ могут опосредовать уничтожение кардиомиоцитов NK-клетками, поэтому данный механизм кардиотоксичности следует учитывать при выборе препарата [19]. Подробная схема, обобщающая патогенез ИКТ-ассоциированного миокардита, представлена на рис. 2.
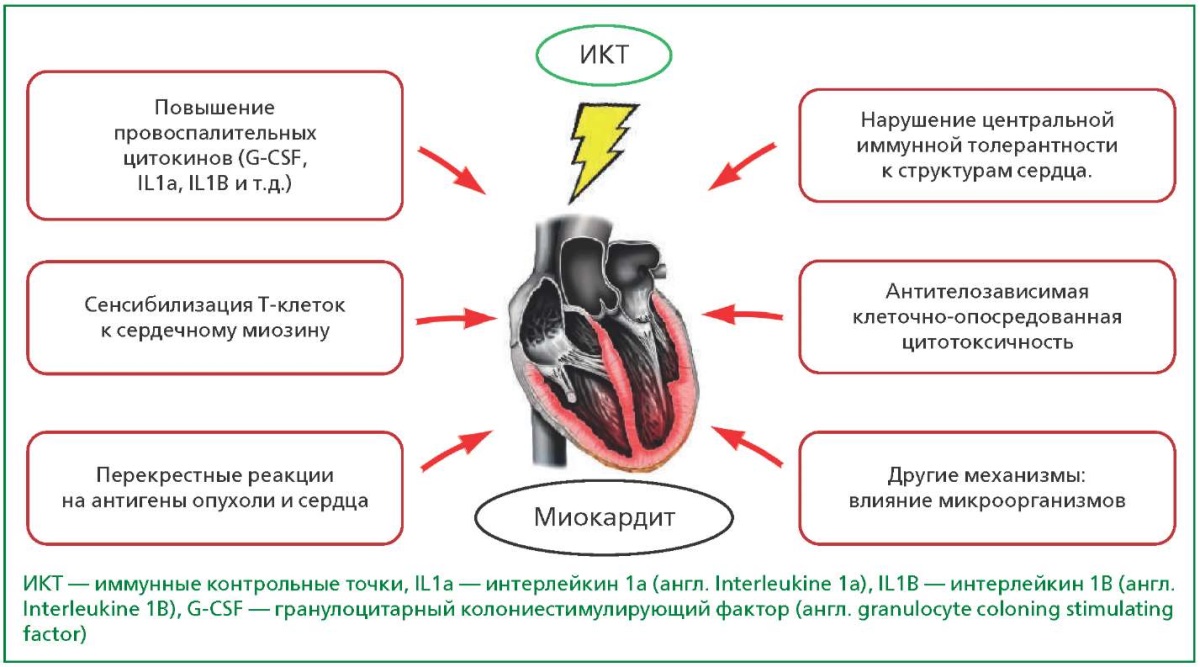
Рисунок 2. Механизмы развития миокардита у онкологических больных,
получающих ингибиторы ИКТ
Дилатационная кардиомиопатия (ДК) — иоНР терапии ингибиторами ИКТ, проявляющаяся дисфункцией и патологическим расширением левого желудочка и чаще всего носящая невоспалительный характер. Относительно механизмов ее возникновения имеется несколько гипотез. Существуют данные, согласно которым ДК, как и любая дисфункция левого желудочка, возникающая при кардиотоксичной противоопухолевой терапии, является прямым следствием опосредованного ингибиторами ИКТ миокардита [9][20]. С другой стороны, имеются экспериментальные исследования, указывающие на вклад в ее патогенез аутоантител [19]. В исследовании H. Nishimura и соавт. было показано, что отсутствие PD-1 вызвало аутоиммунную ДК у мышей с нокаутом гена Pdcd1 и высокими титрами циркулирующих иммуноглобулинов (IgG), отложившихся на поверхности кардиомиоцитов [20]. Последующие эксперименты показали, что аутоантитела вырабатываются против сердечного тропонина I и вызывают сердечную дисфункцию и дилатацию миокарда посредством хронической стимуляции притока ионов кальция в кардиомиоциты [21].
В другом экспериментальном исследовании на мышах, проведенном T. G. Gergely и соавт. непосредственно с блокаторами пути PD-1, было показано, что данные препараты могут быть причиной ДК левого желудочка у мышей C57BL/6J. Так, фракция выброса левого желудочка у таких мышей на фоне использования ингибиторов PD-1 снижалась, а его структура приобретала патологические изменения [22]. Авторами было выделено несколько механизмов развития дилатации. Во-первых, это мог быть повышенный нитрозативный стресс вследствие образования пероксинитрита, вызывающего сердечную дисфункцию. Пероксинитрит вырабатывается с участием никотинамидадениндинуклеотидфосфоксидаза (НАДФН-оксидазы) и синтазы монооксида азота, продуцирующих активные азотистые формы. Ингибиторы PD-1 усиливают экспрессию Nox2, приводя к повышению активности данных ферментов, что в свою очередь и приводит к нитрозависимому стрессу и кардиомиопатии. Во-вторых, еще одним механизмом развития ДК могло быть повышение экспрессии генов провоспалительных цитокинов в тимусе и селезенке. На фоне использования ингибиторов PD-1 у мышей происходило усиление воспалительной передачи сигналов в тимусе, при этом наиболее заметно увеличивался уровень IL-17 — маркера активности Т-клеток. Примечательно, что блокада IL-17 в эксперименте предотвращала сердечную дисфункцию у мышей, что подтверждает роль данного механизма в развитии ДК [23].
Острый коронарный синдром (ОКС) как осложнение терапии ингибиторами ИКТ чаще всего проявляется в виде инфаркта миокарда и реже — нестабильной стенокардией. В настоящее время предложено несколько гипотез, объясняющих патогенез ОКС, первая из них связывает его развитие с атеросклеротическими изменениями коронарных сосудов, вторая — с вазоспастическими эпизодами, третья — с коронарным васкулитом [16].
Атеросклероз — состояние, одновременно являющееся как фактором риска развития различных иоНР, так и непосредственным осложнением терапии ингибиторами ИКТ. В первом случае атеросклеротическая бляшка — источник воспаления и тромбогенных процессов, вызывающий и усугублющий таким образом кардиоваскулярную патологию, в частности ОКС. Во втором же происходит изменение метаболизма холестерина на фоне иммунотерапии [24]. В норме PD-1/ PD-L1 и CTLA-4 сдерживают образование атеросклероза, ограничивая активность Т-клеток, обладающих проатерогенной активностью. В эксперименте на мышиных моделях было показано, что низкие уровни диады PD-1-PD-L1 связаны с более высоким содержанием коронарных атеросклеротических бляшек [25]. Так у мышей PD-L1/2-/-Ldlr-/- развивались более крупные атеросклеротические поражения крупных сосудов с обильным количеством CD8+ Т-клеток и макрофагов. Кроме того, клетки PD-L1/2-/-Ldlr-/- были более восприимчивы к пролиферации, индуцированной АПК, они имели активированный фенотип и экспрессировали более высокие уровни проатеросклеротических цитокинов Т-лимфоцитов: IFN-γ и TNF-а. Эти результаты указывают на критическую роль пути PD-1/PD-L в подавлении проатерогенного ответа Т-клеток и атеросклероза путем ограничения активации Т-клеток, зависимой от антиген-представляющих клеток [25].
Эффекты пути CTLA-4 при атеросклерозе также изучались на модели трансгенных мышей, у которых конститутивная сверхэкспрессия CTLA-4 в Т-клетках уменьшала образование атеросклеротических поражений и накопление внутри бляшек макрофагов и CD4+ Т-клеток [26]. Антитело-опосредованное ингибирование CTLA-4 Ldlr -/- мышей ускоряло прогрессирование атеросклероза, вызывая преимущественно Т-клеточное эндотелиальное воспаление и приводило к 2-кратному увеличению площади бляшек с прогрессирующим, клинически неблагоприятным фенотипом. Аналогичным образом, Т-клеточное воспаление, сосудистая дисфункция и прогрессирование бляшек наблюдались при двойном опосредованном антителами ингибировании CTLA-4 и PD-1 при атеросклерозе у гиперлипидемических мышей (Ldlr-/- и Apoe-/- мышей) [27].
Таким образом, эти экспериментальные модели атеросклероза показали, что пути PD-1-PD-L1 и CTLA-4 уменьшают воспаление, вызванное Т-клетками, тем самым уменьшая развитие и прогрессирование бляшек. Следовательно, их ингибирование в современных схемах иммунотерапии у онкологических больных может активировать Т-клетки в бляшках и усугублять атеросклероз у этих пациентов. Хотя молекулярные пути развития атеросклероза, связанного с ингибиторами ИКТ, помимо PD-1, PD-L1 и CTLA-4, до конца не изучены, в настоящее время исследуются подходы, нацеленные на новые костимулирующие и коингибирующие ИКТ [28].
Другая четко определенная роль в защите от развития атеросклероза связана с Treg-клетками посредством секреции трансформирующего фактора роста β и IL-10, что способствует развитию противовоспалительного фенотипа макрофагов. Исследования также показали, что Treg-клетки конститутивно экспрессируют CTLA-4, и их количество обратно коррелирует с размером и уязвимостью бляшек человека. Кроме того, бляшки пациентов, получавших ингибиторы ИКТ, содержат субпопуляции Т-клеток с маркерами клеточного истощения (высокие уровни PD-1 и перфорина, а также транскрипционные сигнатуры для PDCD1 и LAG-3) и макрофаги с активированными фенотипами, связанными с уязвимостью бляшек [28]. Поскольку истощенные Т-клетки, экспрессирующие PD-1, существуют в атеросклеротических бляшках, это позволяет предположить, что ингибиторы PD-1 могут активировать Т-клетки в бляшках и усугублять атеросклероз.
Васкулит — альтернативное гипотетическое объяснение ОКС, ассоциированного с ингибиторами ИКТ, особенно в случаях отсутствия атеросклероза [29]. Стоит отметить, что васкулит при терапии ингибиторами ИКТ может быть системным и затрагивать не только коронарные, но и другие сосуды. Однако клинически наибольшее значение для формирования ОКС имеет коронарогенный процесс. Предполагаемые патогенетические пути васкулита являются общими относительно всех сосудов и включают в себя повышенные уровни ранее существовавших аутоантител, повышение активности Т-клеток против антигенов, которые присутствуют в здоровых тканях (перекрестные реакции), повышенные уровни цитокинов и усиление воспаления, опосредованного комплементом [28][29]. Высказано предположение, что блокада препаратами PD-L1 в дендритных клетках сосудистой стенки приводит к усилению способности PD-1-позитивных CD4 Т-клеток проникать в стенку сосуда и продуцировать воспалительные цитокины, что приводит к ремоделированию стенки сосуда и его окклюзии. Это способствует беспрепятственному сигналу активации Т-клеток, что создает почву для развития коронарного васкулита [30].
Вазоспазм коронарных артерий — более редкое состояние, однако он может служить этиологическим фактором, приводящим к развитию ОКС [31]. Гипотеза о вазоспазме основывается на накопленном клиническом опыте, зафиксированном в публикациях. Так, в исследовании R. Nykl и соавт. сообщалось о спазме коронарных сосудов с транзиторным подъемом сегмента ST, произошедшем на фоне терапии пембролизумабом при лечении бронхогенной аденокарциномы [32]. В описании клинического случая K. Otsu и соавт., был продемонстрирован пациент, получавший ингибитор ИКТ, на фоне приема которого отмечалась ангинозная боль за грудиной в состоянии покоя [33]. Боль купировалась сублингвальным приемом нитроглицерина, стероидов и иммуномодулирующей терапией. Коронарная ангиография у данного пациента выявила спазм коронарных артерий, который разрешился после интракоронарной инъекции нитроглицерина. При этом у пациента была зафиксирована нормальная эхокардиограмма и не выявлено обструктивной ИБС, что подтверждает связь болевого синдрома с приемом ингибитора ИКТ. В исследовании T. Kumamoto и соавт. была представлена пациентка, принимавшая ниволумаб [34]. У нее не было зафиксировано ангинозных эпизодов в анамнезе, однако на фоне терапии ингибитором ИКТ, появились жалобы на сжимающую, давящую боль за грудиной. При этом боль исчезла вскоре после отмены препарата, что дает основание предполагать прямое влияние ниволумаба на возникновение вазоспастических процессов в коронарном русле.
Аритмии — широкий спектр кардиотоксических иоНР на фоне терапии ингибиторами ИКТ. По характеру они весьма разнообразны и включают нарушения автоматизма (например, синусовую брадикардию) и проводимости, (например, полную атриовентрикулярную блокаду), предсердные аритмии (например, фибрилляцию предсердий), желудочковые аритмии (например, тахиаритмию желудочков) и т.д. [34-36].
Патогенез аритмий представлен несколькими гипотезами. Самая популярная из них предполагает связь с прямой цитотоксичностью Т-клеток, активированных ингибиторами ИКТ [37]. Данная гипотеза основана на гистопатологических исследованиях, выявивших очаговую лимфоцитарную инфильтрацию в синоатриальном и атриовентрикулярном узлах на фоне терапии ингибиторами ИКТ [38]. Аналогичным образом, воспаление проводящей системы Гиса-Пуркинье вследствие прямой цитотоксичности Т-клеток или инфильтрации макрофагов может спровоцировать новые нарушения проводимости, включая блокаду ветвей пучка Гиса или, в тяжелых случаях, задержку внутрижелудочковой проводимости с полной атриовентрикулярной блокадой [16]. Само же кардиотоксическое действие Т-клеток связывается с перекрестными иммунными реакциями. Т-клетки идентифицируют гомологичные антигены опухолевых клеток и кардиомиоцитов и инфильтрируют кардиомиоциты, причем как типичные, так и проводящие, обусловливая нарушение проводимости путем воспалительной реакции [38]. В исследовании A. R. Lyon и соавт. было сделано предположение, что нарушения проводимости, в том числе блокады в системе Гиса-Пуркинье, могут быть опосредованы не только Т-клетками, но и активацией локальных макрофагов за счет усиления продукции цитокинов [37].
Другие гипотезы о генезе нарушений проводимости на фоне терапии ингибиторами ИКТ включают ранее существовавшие сердечно-сосудистые заболевания, воспалительно-фибротические процессы в миокарде, генерализованное воспаление без признаков миокардита, невоспалительную кардиотоксичность, провоцирующую дисфункцию левого желудочка (особенно в сочетании с химиотерапией), воспаление метастазов в миокарде, сопутствующее лечение препаратами, удлиняющими интервал QT [16]. Подчеркивается роль данных факторов у пациентов, получающих лечение анти-PD-1 и анти-PD-L1 препаратами [37], при этом больший эффект анти-PD-1 в отношении аритмий по сравнению анти-CTLA4 был подтвержден в исследовании A. Mascolo и соавт. [39]. Предположительно это связано с тем, что экспрессия PD-1 является конститутивной для миокарда и имеет фундаментальное значение для аутотолерантности, в то время как CTLA-4 имеет большее значение для активности Т-клеток, нежели для прямого предотвращения перекрестных аутоиммунных реакций [26].
Перикардит — иоНР, которая часто сопровождает миокардит, но может возникать и изолированно. При этом перикардит может проявляться как без выпота, так и с ним, повышая риск развития тампонады сердца [32, 40]. Механизмы возникновения перикардита в настоящее время остаются дискуссионными, однако имеются гипотезы, объясняющие его патогенез. Учитывая, что перикардит чаще всего протекает сочетанно с миокардитом, имеются основания полагать, что его возникновение обусловливают механизмы, приводящие к миокардиту [37]. Согласно данной гипотезе, перикард напрямую повреждается Т-клетками, в токсическое повреждение могут вносить вклад цитокины и Т-клеточные лиганды программируемой клеточной гибели, такие как Fas и TRAIL [37]. Подтверждением может стать случай миоперикардита, описанный в работе W. Shalata и соавт. [38].
С другой стороны, существуют гипотезы, предполагающие, что перикардит у пациентов, получающих ингибиторы ИКТ, может быть связан с дополнительной медикаментозной терапией. Согласно исследованию R. K. Paluri и соавт., большинство случаев перикардита на фоне терапии ингибиторами ИКТ связано с наличием рака легких [39]. В свою очередь непропорционально высокая частота перикардита у таких больных объясняется тем, что эти пациенты ранее подвергались лучевой терапии, повреждавшей перикардиальные антигены и приводящей к усилению активности Т-клеток и воспалению. Радиация, таким образом, запускает антиген-специфический эндогенный иммунный ответ, при котором потенциально гомологичные с опухолевыми антигены распознаются Т-клетками, что способствует развитию перикардита [40].
Однако имеются гипотезы, согласно которым кардиотоксичность ингибиторов ИКТ не имеет первичного значения для развития перикардита. Например, потенциальным механизмом перикардита является противоопухолевый ответ на метастатическое поражение перикарда [39]. Согласно исследованиям, этот механизм связан с псевдопрогрессированием микрометастазов в перикарде: после начала лечения обильная Т-клеточная инфильтрация вызывает воспаление и мнимое увеличение размеров опухоли, таким образом, возможно, приводя к перикардиальному выпоту [19]. Вторичный характер перикардит также может иметь при наличии вирусных инфекций или иммуновоспалительных ревматических заболеваний у пациентов, ранее не получавших иммунотерапию. В таком случае блокада ИКТ теоретически также может обострить субклинические проявления уже имеющегося перикардита [40]. Имеются данные и о том, что у некоторых пациентов, ранее перенесших туберкулез, наблюдалась реактивация этого состояния, по-видимому, из-за гиперчувствительности [41].
Некоторые детали о патогенетических механизмах перикардита на фоне терапии ингибиторами ИКТ описываются в работе D. L. Mocan-Hognogi и соавт., где была показана корреляция между перикардитом и полом — мужчины более склонны к кардиотоксическому поражению [42]. Также примечательно, что исследования показывают более выраженную взаимосвязь между заболеванием перикарда и применением терапии анти-PD-1 или анти-PD-L1 по сравнению с терапией анти-CTLA-4 [26]. Исследователи связывают это, в том числе, с высокой активностью популяции макрофагов в перикарде [39]. Работа R. K. Paluri и соавт. подтверждает гипотезу о роли макрофагов, в их исследовании выявлены CD 68+ve клетки в перикардиальной жидкости [43].
Такоцубоподобный синдром (ТПС) — осложнение, проявляющееся острой, часто обратимой апикальной сердечной недостаточностью. В силу его редкости в настоящее время прямая связь между ним и использованием ингибиторов ИКТ остается неопределенной [44]. Причин этому несколько. Во-первых, клинические проявления ТПС могут перекрываться другими формами кардиотоксичности ингибиторов ИКТ (например, миокардитом). Во-вторых, развитие ТПС может быть независимым от терапии опухоли состоянием, обусловленным интенсивным физическим и эмоциональным стрессом у онкологических больных. В-третьих, по данным некоторых исследований имеется ряд стрессоров кардиогенного и некардиогенного происхождения, являющихся потенциальными триггерами ТПС, таких как инфекции, инфузионные реакции, активное курение и сопутствующие сердечно-сосудистые заболевания [45].
Патофизиологические механизмы ИКТ-ассоциированного ТПС остаются в значительной степени неизвестными. Тем не менее, в литературе были представлены различные гипотетические патогенетические механизмы, включающие избыток катехоламинов, коронарный вазоспазм, микрососудистую дисфункцию и активацию определенных сердечных генов [46]. Имеются предположения, что аутореактивные Т-клетки могут перекрестно реагировать с миокардом, вызывая ТПС, ингибиторы ИКТ могут напрямую воздействовать на коронарные артерии, приводя к их спазму, а иммунная блокада может запускать повышенное высвобождение катехоламинов надпочечниками и постганглионарными симпатическими нервами, вызывая тем самым ответ миокарда [47][48]. Подробная схема, обобщающая предполагаемую серию событий при развитии ТПС, представлена на рис. 3.
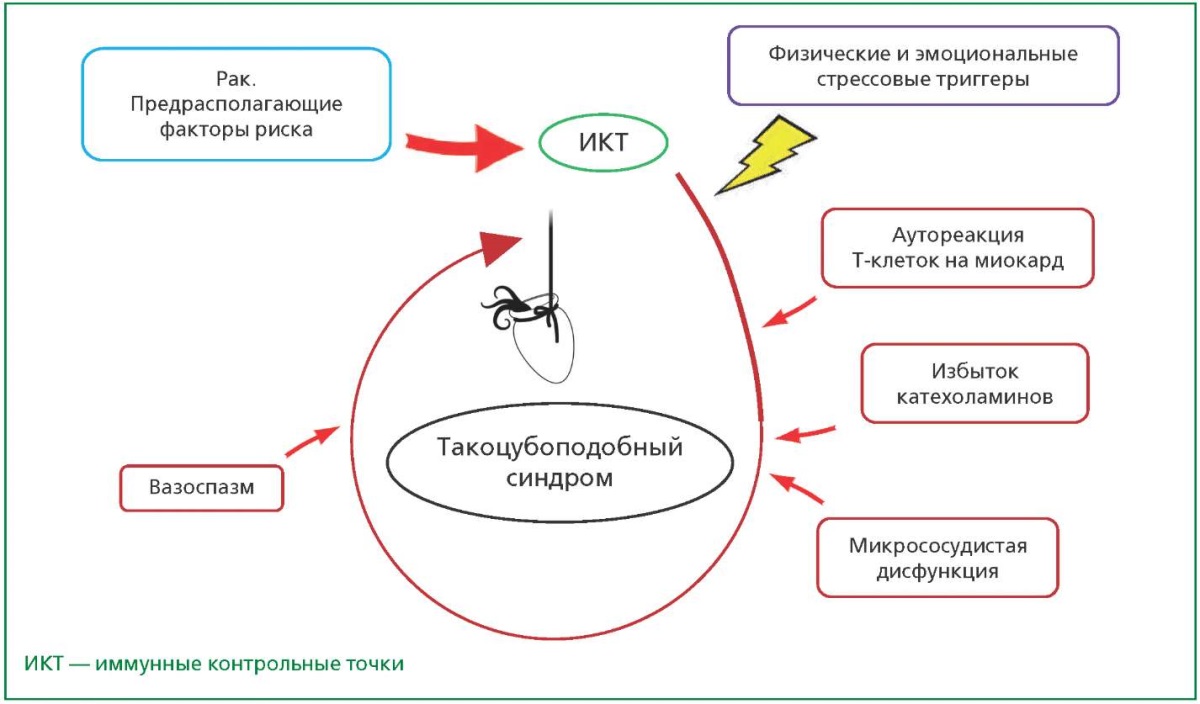
Рисунок 3. Предполагаемые события, приводящие к такоцубоподобному синдрому
у онкологических больных, получающих ингибиторы ИКТ
Кардиотоксичность ингибиторов ИКТ может затрагивать любой отдел сердца, вызывая изменения как воспалительной (миокардит, перикардит, коронарный васкулит), так и невоспалительной природы (аритмии, невоспалительная дисфункция левого желудочка, такоцубоподобный синдром, коронарный вазоспазм и инфаркт миокарда на фоне атеросклероза). За счет разнообразия клинических проявлений при относительной редкости кардиотоксические реакции ингибиторов ИКТ могут создавать трудности при дифференциальной диагностике и лечении онкологических пациентов. Понимание их механизмов повышает возможности специалистов по формированию эффективной стратегии лечения этим классом препаратов с минимизацией риска кардиотоксических осложнений. И хотя при изучении патогенеза иммуноопросредованных нежелательных реакций накоплено достаточно много теоретических, экспериментальных и клинико-эмпирических данных, кардиотоксичность ИКТ представляет собой проблему, требующую дальнейших исследований.
Отношения и Деятельность. Нет.
Relationships and Activities. None.
1. ESC Guidelines on cardio-oncology developed in collaboration with the European Hematology Association (EHA), the European Society for Therapeutic Radiology and Oncology (ESTRO) and the International Cardio-Oncology Society (IC-OS). Eur Heart J. 2022; 43(41):4229-4361. DOI:10.1093/eurheartj/ehac244.
2. Hu J, Tian R, Ma Y, et al. Risk of cardiac adverse events in patients treated with immune checkpoint inhibitor regimens: a systematic review and metaanalysis. Front Oncol. 2021;11:645245. DOI:10.3389/fonc.2021.645245.
3. Chhabra N, Kennedy J. A review of cancer immunotherapy toxicity: immune checkpoint inhibitors. J Med Toxicol. 2021;17(4):411-424. DOI:10.1007/s13181021-00833-8.
4. Tan S, Day D, Nicholls SJ, Segelov E. Immune checkpoint inhibitor therapy in oncology: current uses and future directions: JACC: CardioOncology state-of-the-art review. JACC CardioOncol. 2022;4(5):579-597. DOI:10.1016/j.jaccao.2022.09.004.
5. Franzin R, Netti GS, Spadaccino F, et al. The use of immune checkpoint inhibitors in oncology and the occurrence of AKI: where do we stand? Front Immunol. 2020;11:574271. DOI:10.3389/fimmu.2020.574271.
6. Chocarro L, Blanco E, Zuazo M, et al. Understanding LAG-3 signaling. Int J Mol Sci. 2021;22(10):5282. DOI:10.3390/ijms22105282.
7. Rubio-Infante N, Ramirez-F lores YA, Castillo EC, et al. A systematic review of the mechanisms involved in immune checkpoint inhibitors cardiotoxicity and challenges to improve clinical safety. Front Cell Dev Biol. 2022;10:851032. DOI:10.3389/fcell.2022.851032.
8. Ganesh S, Zhong P, Zhou X. Cardiotoxicity induced by immune checkpoint inhibitor: The complete insight into mechanisms, monitoring, diagnosis, and treatment. Front Cardiovasc Med. 2022;9:997660. DOI:10.3389/fcvm.2022.997660.
9. Wei SC, Meijers WC, Axelrod ML, et al. A genetic mouse model recapitulates immune checkpoint inhibitor–associated myocarditis and supports a mechanismbased therapeutic intervention. Cancer Discov. 2021;11(3):614625. DOI:10.1158/2159-8290.CD-20-0856.
10. Thuny F, Naidoo J, Neilan TG. Cardiovascular complications of immune checkpoint inhibitors for cancer. Eur Heart J. 2022;43(42):4458-4468. DOI:10.1093/eurheartj/ehac456.
11. Moslehi J, Lichtman AH, Sharpe AH, et al. Immune checkpoint inhibitor–associated myocarditis: manifestations and mechanisms. J Clin Invest. 2021;131(5):e145186. DOI:10.1172/JCI145186.
12. Gallegos C, Rottmann D, Nguyen VQ, et al. Myocarditis with checkpoint inhibitor immunotherapy: case report of late gadolinium enhancement on cardiac magnetic resonance with pathology correlate. Eur Heart J Case Rep. 2019;3(1):yty149. DOI:10.1093/ehjcr/yty149.
13. Lim SY, Lee JH, Gide, TN, et al. Circulating cytokines predict immune-r elated toxicity in melanoma patients receiving anti-PD-1–based immunotherapy. Clin Cancer Res. 2019;25(5):1557-1563. DOI:10.1158/1078-0432.CCR-18-2795.
14. Yin J, Yao Z, Pan J, et al. Immune checkpoint inhibitor‐related myocarditis in thymic epithelial tumors: Recent progress and perspectives. MedComm– Oncology. 2023;2(2):e31. DOI:10.1002/mog2.31.
15. Won T, Kalinoski HM, Wood MK, et al. Cardiac myosin-s pecific autoimmune T cells contribute to immune-checkpoint-inhibitor-associated myocarditis. Cell Rep. 2022;41(6):111611. DOI:10.1016/j.celrep.2022.111611.
16. Khunger A, Battel L, Wadhawan A, et al. New insights into mechanisms of immune checkpoint inhibitor-induced cardiovascular toxicity. Curr Oncol Rep. 2020;22(7):65. DOI:10.1007/s11912-020-00925-8.
17. Michel L., Helfrich I., Hendgen-Cotta UB., et al. Targeting early stages of cardiotoxicity from anti-P D1 immune checkpoint inhibitor therapy. Eur Heart J. 2022;43(4):316-329. DOI:10.1093/eurheartj/ehab430.
18. Li X, Peng W, Wu J, et al. Advances in immune checkpoint inhibitors induced-cardiotoxicity. Front Immunol. 2023;14:1130438. DOI:10.3389/fimmu.2023.1130438.
19. Gil-Cruz C, Perez-S hibayama C, De Martin A, et al. Microbiota-derived peptide mimics drive lethal inflammatory cardiomyopathy. Science. 2019; 366(6467):881-886. DOI:10.1126/science.aav3487.
20. Nishimura H, Okazaki T, Tanaka Y, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291(5502):319-322. DOI:10.1126/science.291.5502.319.
21. Okazaki T., Tanaka Y., Nishio R., et al. Autoantibodies against cardiac tro ponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat Med. 2003;9(12):1477-1483. DOI:10.1038/nm955.
22. Gergely TG, Kucsera D, Tóth VE, et al. Characterization of immune check point inhibitor‐induced cardiotoxicity reveals interleukin‐17A as a driver of cardiac dysfunction after anti‐PD‐1 treatment. Br J Pharmacol. 2023;180(6):740-761. DOI:10.1111/bph.15984.
23. Хегай И. М., Трунина И. И., Чеботарева Т. А., и др. Роль иммунной системы в развитии и прогрессировании вирусного повреждения миокарда. Российский вестник перинатологии и педиатрии. 2021;66(3):27-33]. DOI:10.21508/1027-4065-2021-66-3-27-33.
24. Намитоков А. М., Зафираки В. К., Донец Е. К., и др. Агрессивное течение атеросклероза при гиперлипопротеинемии (а): серия клинических случаев. Рациональная Фармакотерапия в Кардиологии. 2023;19(6):591-596]. DOI:10.20996/1819-6446-2023-2873.
25. Bu DX, Tarrio M, Maganto-Garcia E, et al. Impairment of the programmed cell death-1 pathway increases atherosclerotic lesion development and inflammation. Arterioscler Thromb Vasc Biol. 2011;31(5):1100-1107. DOI:10.1161/ATVBAHA.111.224709.
26. Poels K, van Leent MM, Reiche ME, et al. Antibody-mediated inhibition of CTLA4 aggravates atherosclerotic plaque inflammation and progression in hyperlipidemic mice. Cells. 2020;9(9):1987. DOI:10.3390/cells9091987.
27. Suero-Abreu GA, Zanni MV, Neilan TG. Atherosclerosis with immune checkpoint inhibitor therapy: evidence, diagnosis, and management: JACC: CardioOncology State-of-the-Art Review. JACC CardioOncol. 2022;4(5):598615. DOI:10.1016/j.jaccao.2022.11.011.
28. Fernandez DM, Rahman AH, Fernandez NF, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. 2019;25(10):15761588. DOI:10.1038/s41591-019-0590-4.
29. Crout TM, Lennep DS, Kishore S, Majithia V. Systemic vasculitis associated with immune check point inhibition: analysis and review. Curr Rheumatol Rep. 2019;21(6):28. DOI:10.1007/s11926-019-0828-7.
30. Zarifa A, Kim JW, Lopez-Mattei J, et al. Cardiac Toxicities Associated with Immune Checkpoints Inhibitors: Mechanisms, Manifestations and Management. Korean Circ J. 2021;51(7):579-597. DOI:10.4070/kcj.2021.0089.
31. Nykl R, Fischer O, Vykoupil K, et al. A unique reason for coronary spasm causing temporary ST elevation myocardial infarction (inferior STEMI)– systemic inflammatory response syndrome after use of pembrolizumab. Arch Med Sci Atheroscler Dis. 2017;2:e100-e102. DOI:10.5114/amsad.2017.72531.
32. Otsu K, Tajiri K, Sakai S, et al. Vasospastic angina following immune check - point blockade. Eur Heart J. 2020;41(17):1702. DOI:10.1093/eurheartj/ehz796.
33. Kumamoto T, Kawano H, Kurobe M., et al. Vasospastic Angina: An Immune-related Adverse Event. Internal Medicine. 2022;61(13):1983-1986. DOI:10.2169/internalmedicine.8540-21
34. Fonseca M, Cheng E, Do D, et al. Bradyarrhythmias in Cardio-Oncology. South Asian J Cancer. 2021;10(03):195-210. DOI:10.1055/s-0041-1731907.
35. Waliany S, Lee D, Witteles RM, et al. Immune checkpoint inhibitor cardiotoxicity: understanding basic mechanisms and clinical characteristics and finding a cure. Annu Rev Pharmacol Toxicol. 2021;61:113-134. DOI:10.1146/annurev-pharmtox-010919-023451.
36. Johnson DB, Balko JM, Compton ML, et al. Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med. 2016;375(18): 1749-1755. DOI:10.1056/NEJMoa1609214.
37. Lyon AR, Yousaf N, Battisti NML, et al. Immune checkpoint inhibitors and cardiovascular toxicity. Lancet Oncol. 2018;19(9):e447-e458. DOI:10.1016/S1470-2045(18)30457-1.
38. Safi M, Ahmed H, Al-Azab M, et al. PD-1/PDL-1 inhibitors and cardiotoxicity; molecular, etiological and management outlines. J Adv Res. 2021;29:45-54. DOI:10.1016/j.jare.2020.09.006.
39. Mascolo A, Sportiello L, Rafaniello C, et al. Do immune checkpoint inhibitors share the same pharmacological feature in the risk of cardiac arrhythmias? Biomed Pharmacother. 2023;164:114912. DOI:10.1016/j.biopha.2023.114912.
40. Altan M, Toki MI, Gettinger SN, et al. Immune checkpoint inhibitor– associated pericarditis. J Thorac Oncol. 2019;14(6):1102-1108. DOI:10.1016/j.jtho.2019.02.026.
41. Shalata W, Steckbeck R, Abu Salman A, et al. Perimyocarditis Associated with Immune Checkpoint Inhibitors: A Case Report and Review of the Literature. Medicina (Kaunas). 2024;60(2):224. DOI:10.3390/medicina60020224.
42. Mocan-Hognogi DL, Trancǎ S, Farcaş AD, et al. Immune checkpoint inhibitors and the heart. Front Cardiovasc Med. 2021;8:726426. DOI:10.3389/fcvm.2021.726426.
43. Paluri RK, Pulipati Y, Regalla DKR. Immune Checkpoint Inhibitors and Their Cardiovascular Adverse Effects. Oncol Rev. 2023;17:11456. DOI:10.3389/or.2023.11456.
44. Марчев С., Веков Т. Рациональная фармакотерапия при кардио миопатии Такоцубо. Рациональная Фармакотерапия в Кардиологии. 2012;8(6):777-780]. DOI:10.20996/1819-6446-2012-8-6-777-780.
45. Palaskas N, Morgan J, Daigle T, et al. Targeted cancer therapies with pericardial effusions requiring pericardiocentesis focusing on immune checkpoint inhibitors. Am J Cardiol. 2019;123(8):1351-1357. DOI:10.1016/j.amjcard.2019.01.013.
46. Trontzas IP, Vathiotis IA, Kyriakoulis KG, et al; ImmunoTTS Collaborative Group. Takotsubo Cardiomyopathy in Cancer Patients Treated with Immune Checkpoint Inhibitors: A Systematic Review and Meta-S ummary of Included Cases. Cancers (Basel). 2023;15(9):2637. DOI:10.3390/cancers15092637.
47. Guha A, Dey AK, Miller E, et al. Abstract 12013: Trends in Reported Cardiovascular Disease and Hospitalizations in Cancer Patients-Cardio-oncology Patterns Over 14-year From Two Nationally Representative Datas ets. Circulation. 2019;140(Suppl 1):A12013.
48. Polizzotti B. D., Arab S., Kuhn B. Intrapericardial delivery of gelfoam enables the targeted delivery of periostine peptide after myocardial infarction by inducing fibrin clot formation. PLoS One 2012;7: e36788. DOI:10.1371/journal.pone.0036788.
Хидирова Людмила Даудовна
Новосибирск
Лацвиева Анастасия Евгеньевна
Новосибирск
Ведерин Александр Алексеевич
Новосибирск
Хидирова Л.Д., Лацвиева А.Е., Ведерин А.А. Механизмы кардиотоксичности противоопухолевой терапии ингибиторами иммунных контрольных точек: современные достижения. Рациональная Фармакотерапия в Кардиологии. 2024;20(2):265-274. https://doi.org/10.20996/1819-6446-2024-3022. EDN: JNMVKN
Khidirova L.D., Latsvieva A.E., Vederin A.V. Cardiotoxicity mechanisms of antitumor therapy with immune checkpoint inhibitors: new achievements. Rational Pharmacotherapy in Cardiology. 2024;20(2):265-274. (In Russ.) https://doi.org/10.20996/1819-6446-2024-3022. EDN: JNMVKN


Главный редактор
 Драпкина О. М.
Драпкина О. М.
101990, Москва, Петроверигский пер., 10,
Тел.: +7 (499) 553 68 10
е-mail: otsec.rfc@mail.ru








































